
Molecular Bonding Language
Page 1 of 2 • 1, 2 ![]()

Molecular Bonding Language
![]() by Nevyn Thu May 03, 2018 9:06 pm
by Nevyn Thu May 03, 2018 9:06 pm
I have now implemented a parser for it and am in the process of creating a 3D model. I am not currently working with the actual nuclear models. I wanted to get something visual first so that I could confirm the language parser without having to use a debugger and look at the objects in memory. I am creating a 3D network of atoms but only showing the symbol for each atom rather than the nuclear model. It still needs work but everything is created as defined and put into the correct place. Although it is rotating the carousel levels at the moment and I don't want that to happen with this type of view. That will be needed for the actual nuclear models but when using the symbols it just makes it hard to read.
MBL - The Language
The language is meant to be as simple as possible while still allowing some complex molecules to be defined. It is based on element chains where a chain is a series of atoms bonded along the north/south axis. A chain is defined by specifying each atoms symbol, H, He, O, etc, separated by the bonding operator '-'. You can also use whitespace (spaces, tabs, etc) to separate the tokens in an MBL expression. All whitespace is effectively ignored, but it still separates atoms so you can use it if you want to.
An element chain: O-C-Ti-K-O
You do not need to have a bond in an element chain but you must at least have 1 element.
Carousel Bonds
That would allow a lot of molecules to be defined as most bonds are along the north/south axis. However, it is also possible to bond to the carousel positions of an atom. To define that, we use a carousel list which is an array of up to 4 element chains. Each chain in the list corresponds to a carousel position and are in the following order: east, west, front, back. The carousel list is surrounded by square brackets '[' and ']' and each item in the list is comma separated. Note that the carousel list sits right next to the atom that it is bonded to. There is no bonding operator required to separate them.
An atom with carousel bonds in the east and west positions: Fe[O,O]
If we want to have carousel bonds in the front and back but not in the east or west, then we can use the null operator '_' in place of an element chain in the carousel list. Any position can have a null item.
An atom with carousel bonds in the front and back positions: Fe[_,_,O,O]
You can create very complex molecules with this syntax because each item in the carousel list can be an element chain, not just an atom.
Complex carousel bonds: Fe[O-C,Fe[_,O-C]]
Notice that we can nest carousel lists within each other to create branching structures.
Flipping Atoms
In order for this to work, we need a clear definition of the starting orientation of each atoms model. This is irrelevant for a balanced structure but when we have a different number of hook protons in the north compared to the south, we have to know which way the atom is oriented. To accomplish this, all atoms will have their larger hook stack in the south position by default. However, sometimes you need to flip an atom over so that it bonds to the surrounding atoms correctly. In order to accomplish that you use the flip operator '!' before the atom that you want to flip over.
Flipping an atom: O-!Cu-C
Rotating Atoms
You can use the spin operator '*' to rotate an atom about its north/south axis by a specified number of degrees. This will cause all bonds to the carousel positions to also be rotated. You can use decimal values if needed.
Spin an atom: O-Fe*45-C, O-Fe*12.5[H,H]-C
Multiplication
Some molecules have repeating patterns within them and it would be tedious and error prone to define these explicitly all of the time. You can use a multiplication group to repeat an element chain a specified number of times. Multiplication groups are surrounded by '(' and ')' and must be immediately followed by a positive integer number. Note that a multiplication group that is inside of a chain must be preceded by a bonding operator '-' and if it has more elements after the group, then it must also have a following bond operator.
A multiplication group: (O-C)3 -> O-C-O-C-O-C
Within a chain: O-(C-H)2-C-O -> O-C-H-C-H-C-O
You can also nest multiplication groups and they will be evaluated from the inside out.
Nested multiplication group: O-(Fe-(O-Ti)2-O)3 -> O-Fe-O-Ti-O-Ti-O-Fe-O-Ti-O-Ti-O-Fe-O-Ti-O-Ti-O
Cross Bonds
All of the above has been implemented and is working as expected. I have just realised that it can't handle the types of bonds that I have used in my Hydrocarbon Series which have bonds attached orthogonal to other bonds. I will introduce a new grouping structure for these special bonds which will use curly braces '{' and '}' and contain a comma separated list of items. The first item in the list will be a Base Chain. This chain will become a part of the chain that this group is declared in (or it may be the complete chain itself). For each bond in the Base Chain, you can specify 2 more items in the list which represent the Top Chain and Bottom Chain for that bond respectively. You can use the null operator if you don't want anything bonded to a specific location.
A Cross Bond: {C-C,H,H} -> Acetylene
Larger Cross Bond: {C-C-C,He,H,H,He} -> Propene
I changed my mind about the format of these cross bonds. The format above was a bit clunky to work with so I am making it more in-lined. I think it reads better.
A Cross Bond: C{H,H}C -> Acetylene
Larger Cross Bond: C{He,H}C{H,He}C -> Propene
It is also possible to use the bond operator before and after the curly braces if you want to.
e.g. C-{H,H}-C
It can make it look a bit nicer. Readability is very important for any language and even more so here because it is a symbolic language. With familiarity, you should be able to read a simple MBL expression and picture the molecule in your mind.
Out of Scope
With the above language, we can create very complex molecules with little text. Larger molecules will be cumbersome to work with but it is hard to avoid that. Allowing whitespace will help a lot since it allows you to group things without that being a part of the language itself. There are certain structures that will not be handled by this language.
The language is designed to create straight structures with north/south bonds and branches through the carousel bonds. It will not handle ring structures like we find in Hydrocarbons. At least, I can't see a nice way of doing so at the moment, but you never know what the future holds, I guess. If I can find a way then I will attempt it but I feel that a different language might be better suited to that kind of structure.
MBL will also not handle charge field effects like Miles has used for Methane where the H atoms spread apart based on the charge flow through the structure. I might be able to do that eventually, but it requires a level of charge processing that I am nowhere near at the moment.
Ideas and Usage
Feel free to express any ideas that you have about the language or structures that it might support. You can also think about how such a language, or the models that it can generate, could be used. You might even come up with a new way of expressing the language. I am implementing an Atomic Symbol Network at the moment and obviously plan to implement an Atomic Nucleus Network soon. How else might we be able to visualize these structures? What other uses could we put this language to? Think big! Think small! It doesn't matter. All ideas are welcome.
Last edited by Nevyn on Sun May 06, 2018 5:37 am; edited 3 times in total (Reason for editing : Added section of Rotating Atoms)

Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -

Re: Molecular Bonding Language
![]() by Nevyn Fri May 04, 2018 12:41 am
by Nevyn Fri May 04, 2018 12:41 am
Technical Details
Some of you might appreciate some more technical information about what I am creating and how I am going about it. It might also help you to come up with new ideas if you can see the intermediate products that are generated.
Generally, processing a language is broken down into a few tasks and the products of those tasks. The first stage is to tokenize the text which means to break it down into a list of pieces, called tokens, where each token represents something in the language. For MBL, tokens are element symbols, bond, flip, and null operators, group markers and numbers.
If we had the following MBL text:
O-C-Fe[H,H]
Then we would get the following array of tokens:
'O', '-', 'C', 'Fe', '[', 'H', ',', 'H', ']'
Actually, I don't need the bonding operators once it has been tokenized so I really end up with this:
'O', 'C', 'Fe', '[', 'H', ',', 'H', ']'
Now that we have the individual tokens, it is easier to parse the text. All that needs to be done is to iterate through the array and process each token.
Before I do that though, I do a pre-pass that looks for multiplication groups and expands them out into their full form.
MBL: O-(C-Fe)2-O
Tokens: 'O', '(', 'C', 'Fe', ')', '2', 'O'
Expanded: 'O', 'C', 'Fe', 'C', 'Fe', 'O'
That removes a lot of complications for the parsing code.
I won't bore you with the parsing code explanation as I think the product of the parser is of more interest.
Abstract Syntax Tree
The output of a parser is generally called an Abstract Syntax Tree (AST) and it is an intermediate representation of the expression. We now have in-memory data structures, often called objects, that store information from the expression as well as relationships between elements of it. The idea is that the AST will be easier to process in order to generate the final output. Parts of the AST may be raw data as defined in the expression while others may completely remove tokens and replace them with objects to represent them. For MBL, the AST consists of objects called Atoms (strangely enough). The actual result of parsing any MBL expression is a single Atom.
"But, Nevyn, I wrote a full molecule, with lots of atoms, how come you only gave me back 1 of them?"
To answer that, it might help to see exactly what an Atom is, in this context. Here is the Javascript notated structure of an Atom object:
- Code:
{
symbol: "O"
,flip: false
,north: null
,south: null
,east: null
,west: null
,front: null
,back: null
}
The symbol will be used to find the element that this Atom represents. The flip variable is used to flip it over, if needed. The rest are used for bonding to other Atoms.
The returned Atom, called the Root Atom of the molecule, will have the next Atom of the chain stored in its north field. That Atom will have the Root Atom stored in its south field and may have another Atom stored in its north field if the chain keeps going. The east, west, front and back fields are used to store carousel bonded Atoms. So you can see that the complete molecule is represented by the root if you follow the bonds.
Notice that the bonds are bi-directional, they go both ways. This is a convenience but it can lead you into dangerous territory if you are not careful. Essentially, you need to follow the bonds one way only. Since you start with the bottom element, it is best to always move from south to north. If you follow a carousel bond you still move from south to north because those Atoms in the carousel positions are turned sideways such that their south pole will be connected to the carousel hook stacks.
That brings up another important point with respect to flipping an atom. The flip field is only used to turn over the 3D model. It does not affect the bond positions in the Atom object. This means that the north of any atom will be connected to the south of the next atom, regardless of the flipping of that atom. For the carousel levels, the east, west, front or back position would be connected to the south of the atom bonded to it. This keeps things simple and should remove some confusion about how to traverse these Atoms.
Let's look at an example to see how this all fits together. If we defined a simple molecule like this 'O-C-O' then we would end up with 3 Atoms bonded like this:
- Code:
{
symbol: 'O', flip: false
,north: {
symbol: 'C', flip: false
,north: {
symbol: 'O', flip: false
}
}
}
I have omitted the bond fields that are not used in this example for clarity. I have also not included the return bonds as that will be confusing at first because you have to understand the cyclic nature of this data structure. Here is the full structure:
- Code:
{
symbol: 'O', flip: false
,north: {
symbol: 'C', flip: false
,north: {
symbol: 'O', flip: false
,south: {
symbol: 'C', flip: false, ...
}
}
,south: {
symbol: 'O', flip: false, ...
}
}
}
The Atoms in the south positions are not new Atoms. We only have 3 Atoms here and they are referencing each other. Since it is a cyclic structure, I can't actually write it out completely. The '...' represent info that has already been shown and would just lead to a never ending loop. That is why you have to follow the bonds in one direction only.
I am going to have to change that structure a bit since I am about to introduce a new bond type. I'm not sure how to accomplish that at the moment. I might need to introduce a formal Bond object that can store the Atom to be bonded to, the Top Chain and the Bottom Chain of the Bond. It does complicate matters but I am keen to reach a point where I can define most of the molecules that I have modeled in the past through MBL.
Here is how the new Bond object might look:
- Code:
{
to { ... }
,from: { ... }
,top: { ... }
,bottom: { ... }
}
Where the to, from, top and bottom fields are references to Atom objects.
So in the Atom object, the north, south, east, west, front and back fields will contain Bond objects rather than Atoms directly. Using the Bond class in this way will allow me to introduce other bond types in the future just by adding a type field or something similar.
The idea is that the AST can be used for many different purposes. At first I will use it to visualize the molecule but it could be used to perform data processing such as determining the charge flow through the molecule. I'm sure there are other ways to use it too.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Fri May 04, 2018 4:01 am
by Nevyn Fri May 04, 2018 4:01 am
Whitespace
I have implemented whitespace which allows you to replace the bond operator '-' with any whitespace characters. The bond operator was really only there to separate the elements and now that you can use whitespace, it accomplishes the same thing. However, I don't think it looks as nice. The '-' character looks like a bond.
e.g. C-O-C vs C O C
Cross Bonds
Cross bonds are now handled by the parser. I used the Bond object as described above. It complicates traversal of the structure but it allows many more possibilities.
Note the new format. I went with a more in-line structure which can be used like this:
C{H,H}C or C-{H,H}-C or C {H,H} C
or even a mixture of them all.
The 2 items in the list are the top and bottom element chains respectively. These will be bonded to the bond between the last and next atom.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Fri May 04, 2018 6:37 am
by Nevyn Fri May 04, 2018 6:37 am

Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sat May 05, 2018 7:23 am
by Nevyn Sat May 05, 2018 7:23 am
It supports parsing all of the language as described above, but does not show the special cross bonds yet.
I played with showing the atoms as spheres and added a few textures. You can choose them all in the UI of the test page.

The test page also supports parameters on the URL. You can specify the MBL expression, the alignment of the base chain and the type of atom to show. The 'Load' button can be used to take the current settings, put them into a URL and reload the page using that URL. This allows you to easily get a copy of the URL but it also allows you to add molecules to the history of your browser. Play around with a structure using the 'Generate' button and when you are happy, hit the 'Load' button to save it in your history.
e.g. https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=O-C-Fe[O,O]-C-O&align=X&atom=fire
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sat May 05, 2018 5:53 pm
by Nevyn Sat May 05, 2018 5:53 pm
C{H,He}C:

Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 06, 2018 5:14 am
by Nevyn Sun May 06, 2018 5:14 am
Language Update
Atom Rotation
I have added a spin operator '*' that rotates the preceding atom about its north/south axis by the following number of degrees. This allows you to rotate the carousel level and all of its bonds.
e.g. O-Fe*45-C
e.g. O[H,H]-Fe*45[C-H,C-H]-C https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=O[H,H]-Fe*45[C-H,C-H]-C&align=Y&atom=gem02
Last edited by Nevyn on Sun May 06, 2018 5:28 am; edited 1 time in total
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 06, 2018 5:18 am
by Nevyn Sun May 06, 2018 5:18 am
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 06, 2018 8:41 am
by Nevyn Sun May 06, 2018 8:41 am
Language Update
Element Chain Type System
I know I said that ring structures would be out of scope, but it turns out that was just a challenge for myself. I've found a way to express it in the language. I can see how to visualize it with the current text and spheres. I dread having to handle it with nuclear models.
I have introduced a type operator ':' which separates the type information from the chain that it applies to. The first token is the type and any following tokens before the ':' character will be put into an array as parameters. This can be used for various things and I envisage this chain type allowing some special functions to be created.
The first type will be 'ring' and it will probably need a parameter that is the dimension to curl around. You would specify it like this: ring x:(C-O)3.
Any element chain can have a type so they can be used deep inside of the expression and nested. For example: ring x:C-C-C[ring y:(C)5]-C-C.
Another usage for the element chain type that I have thought about is a 'lattice' type. This would allow you to specify 3 parameters which are the number of molecules in each dimension. It would then use the MBL expression to build a molecule which it would clone along each dimension.
You could have a type for 'sheet' that would do the same thing but in 2 dimensions instead of 3. Maybe 'tube' for nanotubes, etc.
The parser can already handle types and their parameters but the visualizer just ignores the information. Using the simpler nodes like spheres and text allows me to do these types of things fairly easily. It may become a nightmare when I get to using the nuclear models.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 06, 2018 11:52 pm
by Nevyn Sun May 06, 2018 11:52 pm
Technical Update
I have made some changes over the last few days and thought it best that I update this technical information to reflect the current status of the project.
Abstract Syntax Tree
As I have added more ways to bond and other information to the MBL language, the AST has to be kept in sync or there is no point in changing the language. This often means that the AST gets more complicated and it certainly has in this case. However, I think the general structure is now faily close to its final form as the current set of classes should be able to handle any future revisions. Although, you never know what the future holds, so they may not.
Atom
The Atom class is used to store all information about an atom that has been specified in the MBL expression. I have only added 1 new property but I have redefined some of the others.
Here is the new structure:
- Code:
{
symbol: <atomic symbol of atom>
,flip: <true or false>
,spin: <degrees to rotate atom about N/S axis>
,north: <null or reference to a Bond object>
,south: <null or reference to a Bond object>
,east: <null or reference to a Chain object>
,west: <null or reference to a Chain object>
,front: <null or reference to a Chain object>
,back: <null or reference to a Chain object>
}
Bond
The Bond class has been introduced to support any special requirements related to the bond between atoms. Specifically, it was created to support cross bonds since they do not really bond to an atom, but to the bond between atoms.
Here is the structure of the Bond class:
- Code:
{
from: <reference to an Atom object>
,to: <reference to an Atom object>
,top: <null or reference to a Chain object>
,bottom: <null or reference to a Chain object>
}
Chain
I have introduced the Chain class to support the new Chain Type mechanism. While it does make traversing the structure a bit more difficult, it actually also makes it much easier because it allowed me to create some functions on the Chain class that would take care of the traversal for you. Given an Atom in the Chain, it will give you the next Atom, for example.
Here is the structure of a Chain:
- Code:
{
type: <null or type of this chain>
,params: <array of strings specific to the type>
,atoms: <array of Atom objects>
}
A Chain contains an array of Atom objects that are a part of that Chain. Generally, you only need the root atom, which is the first in the array. The Chain class has a method, called 'root', that will return you that atom.
The type property, and the params that go with it, are used for special processing. The type field will contain a string that is a single word. The only restriction on the values that the params field can hold is that they can not contain any special characters of the language such as '-', '*', '_', '(', '[', '{', etc. Parameters will generally be simple constructs like numbers, name of a dimension, etc. I may need to introduce an escape mechanism if any special characters are needed in the type or its parameters. The most obvious one is the '-' character which would be needed for negative numbers.
Parser
The result of compiling an MBL expression will now be a Chain object. Previously, it just returned the root Atom of the chain, but now we have chain specific information so it has to return a Chain object.
The general code to compile and traverse a Chain looks like this:
- Code:
...
var chain = MBL.compile( 'some MBL expression' );
var atom = chain.root();
while( atom )
{
// process atom
...
// advance along chain
atom = chain.next( atom );
}
...
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Tue May 08, 2018 6:30 pm
by Nevyn Tue May 08, 2018 6:30 pm
Visualizer Update
Rings
The visualizer can now handle ring structures. They can be a little difficult to work with but a little trial and error should let you get what you want out of them. The main problem is figuring out which way they are going to curl and to help with that you can specify 1 or 2 parameters which dictate the curl axis, the curl direction and the overall orientation of the ring.
Here is a simple ring structure: ring:(C-O-N)3
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=ring:(C-O-N)3&align=Y&atom=fire
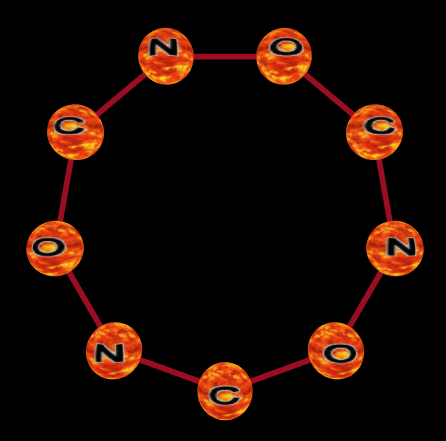
That will give you a default ring containing 3 Carbon atoms, 3 Oxygen atoms and 3 Nitrogen atoms. The chain is oriented in the Y dimension which means that it extends into that dimension. The curl of the ring can be about the X or Z dimension but we can't specify it like that because chains are relative to something else. The user can choose the base chain direction in the UI, but if the ring is inside of another structure then that structure sets its curl axis.
To combat that, I have allowed you to specify the curl axis in a more relative form. You can add a parameter after the 'ring' declaration that sets the curl axis and also the curl direction. The direction is the way that the atoms are placed around the ring. You can think of it as being clockwise or counter-clockwise from a given perspective. However, I have used the same parameter to set both of these things. You can add a parameter with a value of 'east', 'west', 'north' or 'south', although you don't actually need the full word, just the first letter will do.
To figure out which way those are going to curl, you have to put yourself at the base of the chain and imagine that you are looking along it. So you would only really see the first atom in the chain because it extends away from your eye. From that perspective, the ring can move in the north or south direction by curling around the X axis (you are looking down the Z axis) and it can move east or west by curling around the Y axis.
Here is a simple ring structure curling north: ring north:(C-O-N)3
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=ring%20north:(C-O-N)3&align=Y&atom=fire

Here is a simple ring structure curling west: ring w:(C-O-N)3
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=ring%20w:(C-O-N)3&align=Y&atom=fire

Further more, you can actually rotate the complete ring by adding another parameter which is the number of degrees to rotate it about the chain axis (Z axis in the description above).
A simple ring curling west and rotated 45 degrees: ring w 45:(C-O-N)3
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=ring%20w%2045:(C-O-N)3&align=Y&atom=fire

Chain Rotation
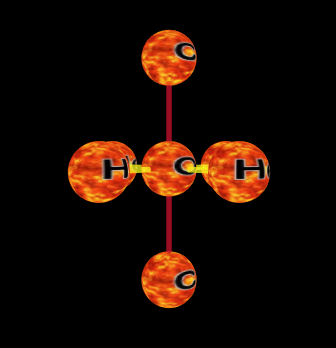
I found the ability to rotate the chain to be quite useful, so I have implemented a way to do the same for a straight chain. You do this by using another chain type called 'rotate' with a parameter that is the number of degrees to rotate around the chain axis. This is only really useful if you have bonds along the chain and will become necessary for very complex molecules.
A rotated chain: rotate 45:C-O[H, H, He, He]-C
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=rotate%2045:C-O[H,%20H,%20He,%20He]-C&align=Y&atom=fire
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Tue May 08, 2018 6:37 pm
by Nevyn Tue May 08, 2018 6:37 pm
MBL: ring e 45:(C)5-O{_, ring e 135:(C)6}(C)5
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=ring%20e%2045:(C)5-O{_,%20ring%20e%20135:(C)6}(C)5&align=Y&atom=fire

Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Jared Magneson Wed May 09, 2018 12:41 am
by Jared Magneson Wed May 09, 2018 12:41 am
Jared Magneson- Posts : 525
Join date : 2016-10-11
Re: Molecular Bonding Language
![]() by Cr6 Wed May 09, 2018 1:39 am
by Cr6 Wed May 09, 2018 1:39 am
https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=O-Cu-[_,O]-Cu-[_,O]-Cu-[_,O]-Mg
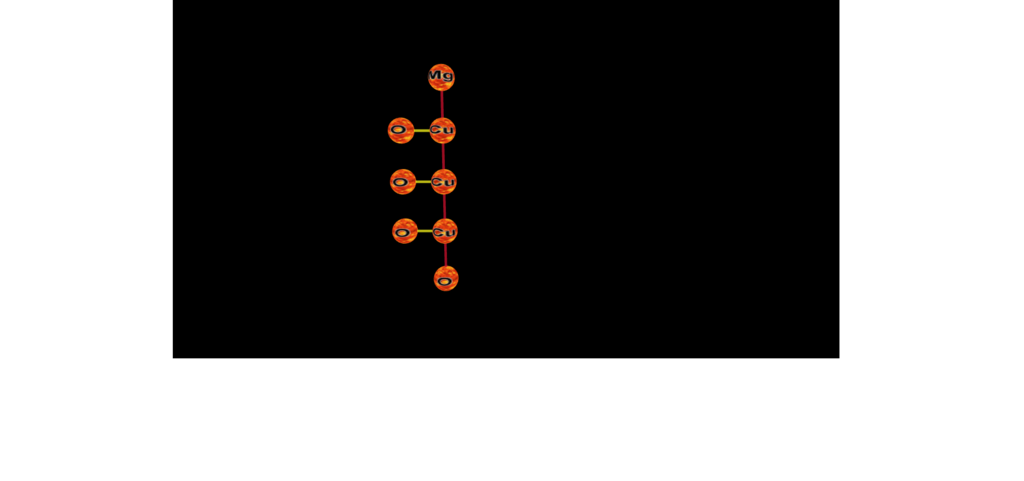
O-O-Cu-[_,_,Cu,Cu]-Mg-O-O (???? possible S.C????)
https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=O-O-Cu-[_,_,Cu,Cu]-Mg-O-O&align=X&atom=fire


or
https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=Cu-[_,_,Cu,Cu]-Mg-O-O-O-O&align=X&atom=fire
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Nevyn Wed May 09, 2018 2:47 am
by Nevyn Wed May 09, 2018 2:47 am
 .
.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Jared Magneson Wed May 09, 2018 2:55 am
by Jared Magneson Wed May 09, 2018 2:55 am
Jared Magneson- Posts : 525
Join date : 2016-10-11
Re: Molecular Bonding Language
![]() by Nevyn Wed May 09, 2018 3:01 am
by Nevyn Wed May 09, 2018 3:01 am
MBL: (O-Cu)2-Mg-O-Cu-O
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=(O-Cu)2-Mg-O-Cu-O&align=X&atom=fire

Super-conductors are pretty boring as far as molecular structure goes. Straight chains as far as I can tell. You're looking for that straight path through all of the atoms. The red bond lines actually represent the through-charge of the molecule. That is the transmission line that we force charge through.
Although it may be better to have a copper on the bottom/start of the molecule as it has 2 protons in its south pole. This gives an imbalance N/S that gives the through-charge a direction to flow. I don't think that is essential but it may help.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Wed May 09, 2018 8:29 pm
by Nevyn Wed May 09, 2018 8:29 pm
This is easy enough for a straight chain, which is what I was playing with last night. Rings, on the other hand, are a real problem. For the sphere or text, I know the length of each atom (or the length that I want it to have) and I can find the radius of the ring easily with a bit of trigonometry. Now I have to create a ring with different sized pieces and I don't know how to do it.
My only thought at the moment is to average the lengths of all atoms in the ring and use that in the same way that I have built the sphere based rings. This may work for some cases, but I fear that it will fail spectacularly at some stage. I will give this a try and see how it looks but I am on the look out for another way.
This seems like it will require some esoteric math, but I was hoping some of you might have some idea on how to do this or where to look for more information.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by LongtimeAirman Wed May 09, 2018 11:19 pm
by LongtimeAirman Wed May 09, 2018 11:19 pm

Hey Nevyn, great work! I really don't know if this is the info you're requesting but I wanted to give you a quick answer before l quit for the day. I believe you are trying to solve cyclic irregular convex polygons. Each unequal length atom is a different polygon side, all the atomic connections lie on a circle. Notice all the radii are the same, if I'm not mistaken, the problem becomes finding the internal angles. Try searching on that term, when I did I ended up with the image above and paper below.
On Circumradius Equations of Cyclic Polygons
https://bib.irb.hr/datoteka/402976.main1.pdf
by Dragutin Svrtan
June 5, 2009
1 Introduction
Cyclic polygons are the polygons inscribed in a circle. In terms of their side lengths a1,a2,...,an, their area S and circumradius r are given in case of triangles and quadrilaterals explicitly by the following well known formulas: the Heron’s formula ( 60 B.C.) for the area and the circumradius r of triangles (by letting A = (4S)2,ρ = 1/r2 ): A−(a+b+c)(a+b−c)(a−b+c)(−a+b+c) = 0 a2b2c2ρ−(a+b+c)(a+b−c)(a−b+c)(−a+b+c) = 0
Hope this helps.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Re: Molecular Bonding Language
![]() by Nevyn Wed May 09, 2018 11:55 pm
by Nevyn Wed May 09, 2018 11:55 pm
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Cr6 Thu May 10, 2018 2:13 am
by Cr6 Thu May 10, 2018 2:13 am
Also, as we know the proton generally "rules" in the Mathis' papers. What could be readily applied as "rules" to form/not form certain structures according to the CF flows? I see how Cu3MgO4 could be rendered in a multitude of ways...but which ones could be ranked by probable formation -- like the straight line bonds which does look better than others. I guess I'm looking for a "look up" rule for bonds to prohibit forms that could simply not be created. The tool already can do this pretty well by just rendering "gaps" in the links -- and I ended up rewriting a few MBL "forms" with this approach. It didn't look right...like immediately -- and this is good. If a certain molecule syntax is thrown to the MBL rendering engine... which might be the best forms to show via Mathis? I know this extremely speculative... but even a few rules might be better than none. A lot to code through though....like with a call to a database or a lengthy parsing function. I figure that you guys (Neyvn, LTAM, Jared, Ciaolo) have already thought about it as well...rules to allow more automation with molecule formations?
Here's Miles in the Solid Light paper:
Miles Mathis wrote:
Well, in a sense, we don't. Superconduction turns out to be a bit of a misnomer. Without nuclear spin,
the nucleus is no longer conducting at all, rigorously. It is only continuing to provide a path, given by
the nuclear structure, but the nuclear vortices are gone. The nucleus is no longer driving charge
through, it is now only allowing charge through. The driving force of the conduction must be supplied
by the incoming current itself. Remember, a superconductor is providing no resistance to a given
charge stream or ion stream. But we have to supply the current from outside. A superconductor can't
create its own current from an unstructured external field, as a normal conductor can. A
superconductor can only provide a zero-resistant path for a pre-existing structured field.
In this line, we also have to remember that although supercold can nullify the overall nuclear spin, it
won't nullify the spins on all the protons in the nucleus. If we apply the external current to our
superconducting material, properly aligned, that current alone will spin the protons it contacts on the
nuclear poles. Think of the fan again. A moving fan will pull in wind from behind, but the reverse is
also true. If you apply wind from behind a non-moving fan, the wind will start the fan turning. So
although the carousel levels aren't spinning in a superconductor, the protons on the poles are. We could
then call this conduction, if we like, since the spinning protons then conduct the charge through.
Strictly, we would have to say the incoming charge (current) is the cause of the spin, rather than the
spin being the cause of the charge channeling, but you see what I mean.
As usual, there are no real pulls in my physics, but the field densities here create potentials that give us the
appearance of pulls. Again, it helps to think of the protons as fans that blow photons in certain directions. Just
as fans seem to pull wind as well as push it, so do the spinning protons and alphas.
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Nevyn Thu May 10, 2018 8:15 am
by Nevyn Thu May 10, 2018 8:15 am
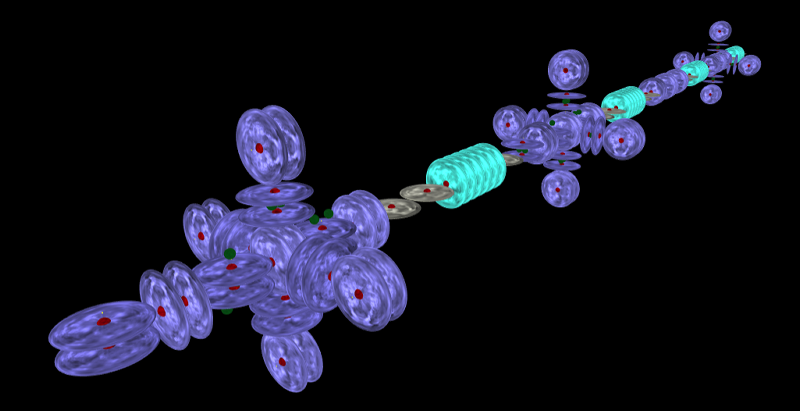
MBL: Cu-O-Cu-O-Mg-O-Cu-O

Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Thu May 10, 2018 8:38 am
by Nevyn Thu May 10, 2018 8:38 am
Regarding the rules of building molecules, it is difficult to say. There are many rules, or guidelines, but they are applicable in different scenarios. Super-conductors like straight paths, aromatic hydro-carbons like rings. To get a feel for it, you can look at some known molecules that do not contain many atoms. Two or three. Have a look at their models and see how they might fit together. Then take your best candidates and look for charge flow characteristics. Look at the known properties of the molecules and find reasons for those properties.
There are some laws that can not be broken. You can't force more charge into an atom than its interior can handle. This limits the size of proton stacks in bond locations although it is not always a consideration. The outer most stacks determine the charge flow so internal bonds might use larger stacks but they will not conduct at full power.
Are you thinking of a molecule generator? An app that, given the rules you want to apply, will give you potential molecules that satisfy those rules? You might need to give it a list of atoms that can be used. That would be a pretty good app.
Another way I can interpret your question is to create code that validates the MBL given a set of known rules. That is feasible, to some extent. Now that I am working with the nuclear models, I have the data for each atom at my disposal. This allows me to look into the atom and determine if a bond is feasible.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by LongtimeAirman Thu May 10, 2018 10:25 am
by LongtimeAirman Thu May 10, 2018 10:25 am
Cyclic polygons. One more paper - in a free e-book, Advances in Discrete Differential Geometry. I was worried whether the cyclic polygons solution must exist, this paper seems to prove they do.
https://link.springer.com/chapter/10.1007/978-3-662-50447-5_5
Advances in Discrete Differential Geometry pp 177-195 | Cite as
A Variational Principle for Cyclic Polygons with Prescribed Edge Lengths
Abstract. We provide a new proof of the elementary geometric theorem on the existence and uniqueness of cyclic polygons with prescribed side lengths. The proof is based on a variational principle involving the central angles of the polygon as variables. The uniqueness follows from the concavity of the target function. The existence proof relies on a fundamental inequality of information theory. We also provide proofs for the corresponding theorems of spherical and hyperbolic geometry (and, as a byproduct, in 1+11+1 spacetime). The spherical theorem is reduced to the Euclidean one. ... .
////////////////////////////////////////////////
https://pdfs.semanticscholar.org/38db/8940901febf1aef7bac87305b5631d84e639.pdf
CYCLIC POLYGONS IN CLASSICAL GEOMETRY
REN GUO AND NILGUN SONMEZ
Abstract. Formulas about the side lengths, diagonal lengths or radius of the circumcircle of a cyclic polygon in Euclidean geometry, hyperbolic geometry or spherical geometry can be unified.
////////////////////////////////////////////////
The abstracts do read weird at times. Ok, I'll quit unless I find something better.
.
Last edited by LongtimeAirman on Thu May 10, 2018 5:30 pm; edited 2 times in total (Reason for editing : added second paper)
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Re: Molecular Bonding Language
![]() by Nevyn Thu May 10, 2018 7:09 pm
by Nevyn Thu May 10, 2018 7:09 pm
I just used the average length of each atom in the ring to calculate the ring radius and it worked reasonably well. As far as I am aware, rings are only made with small atoms like carbon or oxygen, maybe some nitrogen occasionally. But I'm not an expert on all molecules, so there may be some larger ones. I imagine that any atom with a carousel level is not likely to be in a ring.
It still needs more work before I publish it to the web but I am impressed so far. It really brings the chains to life. What was a boring straight chain yesterday, looks awesome today!
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Thu May 10, 2018 7:13 pm
by Nevyn Thu May 10, 2018 7:13 pm
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 13, 2018 5:48 am
by Nevyn Sun May 13, 2018 5:48 am
Visualizer Update
Atomic Models
I have implemented a new visualizer that uses the atomic models from AtomicViewer, complete with charge emission. It supports the full language which means that it does allow you to create illegal structures. When you get to see the full nuclei, most bad configurations will be obvious.
The placement of the atoms needs some thought. I think that the emissions of the hook stacks should be overlapping in a bond. Now that I have everything running, I can look into the finer details like this. Happy to hear anyone's thoughts too.
I'm really happy with the models. Even simple molecules look awesome. I will add some controls to enable/disable the charge emission shaders. They look great, but get in the way when you are thinking about chemistry. They also don't photograph very well. Much like real charge!

Last edited by Nevyn on Sun May 13, 2018 8:36 am; edited 1 time in total (Reason for editing : Added screenshot)
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by LongtimeAirman Sun May 13, 2018 1:16 pm
by LongtimeAirman Sun May 13, 2018 1:16 pm
Hey Nevyn, please don't hate me because Euclid made the problem intractable.
Constructing a cyclic polygon given the edge lengths.
https://chaosinmotion.blog/2016/10/02/constructing-a-cyclic-polygon-given-the-edge-lengths/
Posted on October 2, 2016
.The author wrote. So I have a problem: given the length of the edges L = {l0, l1, … lN} of an N-sided irregular polygon with N > 3, I need to construct the values for the radius R and the angles A = {a0, a1, … aN} such that the points P = { R, ai } form a closed polygon with the lengths given.
I searched through the internet and found all sorts of articles on the subject, https://en.wikipedia.org/wiki/Circumscribed_circle but a day of searching and I was unable to find a way that I could construct the values R and A. I’m sure it’s out there, but I figured it’d be a good exercise to do it myself.
Based on my reading apparently there is no current solution to the problem–so I went ahead and built a simple algorithm to construct the values. The idea is outlined below.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Re: Molecular Bonding Language
![]() by Nevyn Sun May 13, 2018 4:31 pm
by Nevyn Sun May 13, 2018 4:31 pm
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Cr6 Sun May 13, 2018 5:42 pm
by Cr6 Sun May 13, 2018 5:42 pm
It's fun too after imagining the bonding and then seeing it modeled in the MBL. Nicely done!
I was just thinking of having simple slide-in/out type "molecular label" even as top layer....something that if someone took a screenshot it could show on the image if they needed it.
Nice Work! Imagination realized!
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Cr6 Sun May 13, 2018 6:17 pm
by Cr6 Sun May 13, 2018 6:17 pm
Nevyn wrote:How's this for Cu3MgO4?
MBL: Cu-O-Cu-O-Mg-O-Cu-O
Looks very similar to this one Zn3MgO4:

MBL: Zn-O-Zn-O-Zn-O-Mg-O
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 13, 2018 7:02 pm
by Nevyn Sun May 13, 2018 7:02 pm
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 13, 2018 7:07 pm
by Nevyn Sun May 13, 2018 7:07 pm
It would be interesting if we start looking at the known super-conductors and seeing what atoms can be swapped with each other and then see if they end up being found as new super-conductors. That would be a great prediction for Miles theories.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sun May 13, 2018 8:52 pm
by Nevyn Sun May 13, 2018 8:52 pm
Language Update
Cross Bonds in a Ring
While playing around with the ring structures, it became apparent that the cross bonds were not dealing with the last atom bonding back to the first atom. This doesn't apply to straight chains so I didn't need to think about it before. Now, we need to be able to specify the last cross bond, even though it doesn't look like it bonds to anything.
To support this, you can specify a cross bond as the last item in a chain or in a multiplication group. It will only be used in a ring and straight chains will ignore it since it will not have anything to bond to.
e.g. MBL: ring:C{H,H}C{H,H}C{H,H}C{H,H}C{H,H}
or more succinctly: ring:(C{H,H})5
Visualiser Bug
I have found that there is a bug in the way cross bonds are handled for the nuclear models. They are not being positioned correctly. They seemed fine last night when I was working on it, but I checked them on my site this morning and they are not working. They do in some directions but not in all.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Tue May 15, 2018 5:45 am
by Nevyn Tue May 15, 2018 5:45 am
Bug Fix
I fixed the problem with the cross bonds and in doing so, I have stumbled upon an interesting result that I am going to keep. When you use a ring and define whether it is N/S or E/W then the cross bonds will orient themselves in different directions to the ring. The E/W ring will have the cross bonds pointing into and out of the ring while the N/S ring will point them out to the sides. I think it is a useful feature so it can stay.
East ring with cross bonds:

West ring with cross bonds:

Notice that the atoms in the cross bond have switched between inside and outside of the ring.
North ring with cross bonds:

The south ring does not change the cross bonds from the north ring.
UI Update
Charge Controls
I have added some checkboxes to enable/disable charge particles and the discs. They may seem a bit strange in how they work but it is kind of useful at the same time. If you generate a model while all charge shaders are turned off (everything except the disc is disabled) then the shaders will not even be created. This helps to reduce performance requirements and run better on some devices. However, when any of them are turned on, then you can turn them off instantly. No need to regenerate to apply the changes. It is just making them invisible so it may not reduce computational load but it is handy for quickly showing and hiding the charge particles.
Remember that you have to generate the model (cog icon) to apply most settings, including turning on the charge shaders.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Tue May 15, 2018 5:49 am
by Nevyn Tue May 15, 2018 5:49 am
Any thoughts?
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Tue May 15, 2018 6:07 am
by Nevyn Tue May 15, 2018 6:07 am

Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Thu May 17, 2018 5:49 am
by Nevyn Thu May 17, 2018 5:49 am
UI Update
Image Capture
I have fixed the image capture feature so that it renders the full scene as seen on screen. Without the white borders around it. I have also implemented an image properties dialog box that allows you to select the resolution of the rendered image: 720, 1080 or 4K. The dialog box will show when you press the capture icon. It will also remember your settings until a page refresh.
I tried to get a label drawn to the image after the 3D engine has rendered the molecule but it turns out I can't draw in 2D after the canvas has been used for 3D. I may be able to find another way around it.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Cr6 Sat May 19, 2018 1:53 am
by Cr6 Sat May 19, 2018 1:53 am
 . Met an engineer at a bar and drifted into talking about Miles and the MBL. Heard the phrase: "What am I looking at, no really what is this?" several times as he typed in his own molecules. Thinking people definitely get a twinge of "hmmmm". The engineer bought me a free scotch after explaining bits of Mathis' site and yours and "ours" (Forumotion)... now if MBL could only be used to flirt with nice ladies...
. Met an engineer at a bar and drifted into talking about Miles and the MBL. Heard the phrase: "What am I looking at, no really what is this?" several times as he typed in his own molecules. Thinking people definitely get a twinge of "hmmmm". The engineer bought me a free scotch after explaining bits of Mathis' site and yours and "ours" (Forumotion)... now if MBL could only be used to flirt with nice ladies...
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Nevyn Sat May 19, 2018 2:56 am
by Nevyn Sat May 19, 2018 2:56 am
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Sat May 19, 2018 3:08 am
by Nevyn Sat May 19, 2018 3:08 am
Official Release
I have officially released the MBL Language and Renderer, which basically means I added it to my front page. Oh, and I created a formal specification of the language as well as a cheat-sheet type guide for quick reference.
https://www.nevyns-lab.com/mathis/app/mbl/
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Cr6 Sun May 20, 2018 3:09 am
by Cr6 Sun May 20, 2018 3:09 am
Now if it could only be made into an Apple/Android app... then you could get residuals.
Based on Apache Cordova:
https://build.phonegap.com/
https://github.com/phonegap/
https://crosswalk-project.org/blog/crosswalk-final-release.html
https://twitter.com/xwalk_project
https://crosswalk-project.org/documentation/about/demos.html
http://tmtg.net/glesjs/
https://github.com/jeremy-brenner/cordova-threejs
https://blog.ludei.com/tag/threejs/
https://stackoverflow.com/questions/40802820/how-to-add-crosswalk-webview-in-my-own-android-library-module/40858878#40858878
For fun:
Physijs
Physics plugin for Three.js
https://chandlerprall.github.io/Physijs/
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Cr6 Sun May 20, 2018 9:34 pm
by Cr6 Sun May 20, 2018 9:34 pm
Gd in the frustrated antiferromagnet Gd3Ga5O12.
https://warwick.ac.uk/fac/sci/physics/research/condensedmatt/supermag/
Hidden order in spin-liquid Gd3Ga5O12
http://science.sciencemag.org/content/350/6257/179
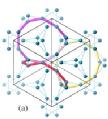
Gd3Ga5O12(?)
https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=O-Gd-O-Ga-O-Gd-O-Ga-O-Gd-O-Ga-O-O-Ga-O-O-Ga-O-O-Ga
Gd2Sn2O7(?)
https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=(O-Gd-O-Sn-O)2-O
Just wanted to ask if there is a way to pre-parse current Chem notation into the MBL?
Like a pre-parse field for MBL, that just lays it all out on a long line?
I was thinking of building one in SQL to re-order the notation to a more friendly MBL format. Basically it would parse the elements, then then numbers and then add the elements back using ((element +'-' ) * (#-1)) or Multiplication Groups (or something like this). I know there are a lot of specific case details but it would be kind of useful to get things laid out to allow easier cut/paste for traditional Chem notation... thoughts? Not looking for the layout to be perfect but just for all elements to be accounted for.
The reason is I have a bunch of molecules in a SQL database but they are in the typical format of Gd2Sn2O7 that would be cool to transform with a click into something like "(O-Gd-O-Sn-O)2-O" or "Gd-Gd-Sn-Sn-(O)7" that if looked good could then be rendered in the MBL viewer?
https://www.nevyns-lab.com/mathis/app/mbl/spec.php#elements
Abstract
Despite the availability of a spin Hamiltonian for the Gd3Ga5O12 garnet (GGG) for over twenty five years, there has so far been little theoretical insight regarding the many unusual low temperature properties of GGG. Here we investigate GGG in zero magnetic field using mean-field theory. We reproduce the spin liquid-like correlations and, most importantly, explain the positions of the sharp peaks seen in powder neutron diffraction experiments. We show that it is crucial to treat accurately the long-range nature of the magnetic dipolar interactions to allow for a determination of the small exchange energy scales involved in the selection of the experimental ordering wave vector. Our results show that the incommensurate order in GGG is classical in nature, intrinsic to the microscopic spin Hamiltonian and not caused by weak disorder.
https://www.researchgate.net/publication/1847995_Spin_Hamiltonian_Competing_Small_Energy_Scales_and_Incommensurate_Long_Range_Order_in_the_Highly_Frustrated_Gd3Ga5O12_Garnet_Antiferromagnet
Low temperature magnetic properties of geometrically frustrated Gd2Sn2O7 and Gd2Ti2O7
http://iopscience.iop.org/article/10.1088/0953-8984/15/45/016/meta
http://iopscience.iop.org/article/10.1088/0953-8984/15/45/016/pdf
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Nevyn Mon May 21, 2018 1:18 am
by Nevyn Mon May 21, 2018 1:18 am
What we really want is a pre-parser followed by a best attempt at arranging the atoms based on the hook proton stack sizes. We might be able to create profiles for various molecule types. One for super-conductors, one for hydrocarbons, one for rings, etc. Maybe some questions like 'Is it a conductor or magnetic?'. You can already see the complexity in that endeavor. I think we need to identify those profiles and the rules that they contain to see how this might work.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Mon May 21, 2018 3:06 am
by Nevyn Mon May 21, 2018 3:06 am
I had a look over the code to see what I could do to support standard chemical notation when it dawned on me that I could possibly handle it in the MBL language itself. I dove into the MBL parser to see how that might work when I realised that it didn't need to go in the parser but could be handled in the tokenizer. All I had to do was look for upper and lower case characters to determine if I was in an element declaration and to watch for numbers immediately following an element. Since MBL also uses ( and ) for multiplication, they fall through without any hassles.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Mon May 21, 2018 3:16 am
by Nevyn Mon May 21, 2018 3:16 am
Language Update
Standard Chemical Notation
MBL now supports the usage of standard chemical notation such as MeBr, O2, Cu(OC)2.
They will only create straight chains and multipliers are just expanded in-place. The order is the same as you put them in the expression.
Visualizer Update
Nuclear Models
I have made various changes to the nuclear model code to support the bonded atoms such as Uranium. I changed our JSON format to do so. It now supports 2 different kinds of atoms, the standard levels and these multi-nuclei atoms. I have modeled from Uranium up to Cerium.
You can use them in your MBL but they have problems. Mainly around the carousel levels. Since these atoms actually have 2 atoms inside of them, they have 2 carousel levels. The placement code in the Renderer can't support that at the moment but you can always just use the root atoms in your MBL in place of these bonded atoms so that you can place bonds onto the carousel of each part.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Nevyn Mon May 21, 2018 9:21 am
by Nevyn Mon May 21, 2018 9:21 am
Language Update
Multiple Carousel Levels
I have added support for an atom with multiple carousel levels. You just add another carousel list after the first and they are applied from bottom to top within the atom.
MBL: U[CC,CC,CO,CO][CC,CC,CO,CO]
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=U[CC,CC,CO,CO][CC,CC,CO,CO]&align=Y&atom=nucleus

This is probably a waste of time because you can't really bond to these locations. The main charge flow is down the N/S axis and there is not enough carousel output to support a bond. It does look cool though!
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Re: Molecular Bonding Language
![]() by Cr6 Fri May 25, 2018 1:49 am
by Cr6 Fri May 25, 2018 1:49 am
Nevyn wrote:Cr6, it must be your birthday!
I had a look over the code to see what I could do to support standard chemical notation when it dawned on me that I could possibly handle it in the MBL language itself. I dove into the MBL parser to see how that might work when I realised that it didn't need to go in the parser but could be handled in the tokenizer. All I had to do was look for upper and lower case characters to determine if I was in an element declaration and to watch for numbers immediately following an element. Since MBL also uses ( and ) for multiplication, they fall through without any hassles.
Hi Nevyn,
Sorry for the delay on the response. I must say that earlier this month was my birthday and that your javascript app was the best present I have ever received aside from a few gifts from my mother and father as a wee child (the updated '77 version of Rocke'm Socke'm Robots comes to mind for some reason...) The "tokenizer" was a left hook when I first used it.

https://en.wikipedia.org/wiki/Rock_'Em_Sock_'Em_Robots
God Bless the MBL...and all your work... it is really cool!!!
Last edited by Cr6 on Sun May 27, 2018 1:23 am; edited 1 time in total (Reason for editing : Corrected typo-bad grammar in a half-drunken excited post...)
Cr6- Admin
- Posts : 1178
Join date : 2014-08-09 -
Re: Molecular Bonding Language
![]() by Nevyn Sat May 26, 2018 12:12 am
by Nevyn Sat May 26, 2018 12:12 am
Language Update
Extending a Molecule
I have added 2 more Chain Commands that allow you to extend a chain by copying it and stamping it a given number of times.
The line command can be used to extend a straight chain. You can build sheets by using a multiplication group in the expression.
The line command can accept 2 parameters. The first is the number of times to copy the chain and is a positive integer > 0. The second parameter can be a direction north, south, east or west, or just the first letter. This is used to orient the chain before it is copied.
MBL: line 6:(C{C,C}C)10
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=line%206:(C{C,C}C)10&align=X&atom=nucleus

The tube command can be used to extend a ring. It can accept 2 parameters. The first is the number of times to copy the ring and is a positive integer > 0. The second parameter can be a direction. Only east and west make sense for a ring since it is restricted in the dimensions that it can grow into. However, the renderer allows you to use north and south as synonyms for east and west respectively.
MBL: tube 6 n:(C{C,C}C)10
URL: https://www.nevyns-lab.com/mathis/app/mbl/mbl.html?mbl=tube%206%20n:(C{C,C}C)10&align=X&atom=nucleus

Even though it looks like everything is bonded together, it is really just a copy of each chain placed next to each other. It is up to you to ensure that the bond points match and make sense.
You will get a performance hit if you use too many atoms. I have disabled transparency on the proton stacks to help. You can turn on the charge emission shaders and it looks fantastic but you will need decent graphics hardware to render it.
Nevyn- Admin
- Posts : 1887
Join date : 2014-09-11
Location : Australia -
Page 1 of 2 • 1, 2 ![]()
» Atomic Modeling Language
» Spinning Particle Language
» Natural Language Processing (NLP) and newer algorithms
» Mass Defect May Help Explain Bonding & Gravity