
Miles Periodic Table with Standard Periodic Table reference
Page 12 of 13 •  1, 2, 3 ... , 11, 12, 13
1, 2, 3 ... , 11, 12, 13 ![]()
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Fri Apr 19, 2024 6:44 pm
by Chromium6 Fri Apr 19, 2024 6:44 pm
Here's the update needed.Might work.
CREATE OR ALTER PROCEDURE [dbo].[spBuildAtomicMilesMathisOrbitalsDetailAllBonds]
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Fri Apr 19, 2024 9:52 pm
by LongtimeAirman Fri Apr 19, 2024 9:52 pm
Ok, replaced CREATE PROCEDURE with CREATE OR ALTER PROCEDURE.
- Code:
...
-- CREATE PROCEDURE [dbo].[spBuildAtomicMilesMathisOrbitalsDetailAllBonds]
CREATE OR ALTER PROCEDURE [dbo].[spBuildAtomicMilesMathisOrbitalsDetailAllBonds]
...
- Code:
Commands completed successfully.
Completion time: 2024-04-19T18:42:24.0864282-07:00
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Mon Apr 22, 2024 6:22 pm
by LongtimeAirman Mon Apr 22, 2024 6:22 pm

Above, we see Miles’ charge field Germanium (Ge 32) atom on the left, showing blue single alphas (two protons each) and (black) single protons. The ellipses indicate each proton or proton stack’s charge emission plane. From Miles’ paper “PERIOD FOUR of the Periodic Table”, as well as more recently in “The Rule of Four”, and “Fractional Quantum Hall Effect”, along with Arlo Emerson’s Molybdenum, (Mo 42). “reds are double alphas”, nine slots each containing 3 protons, a period 5 atom compared to Germainium’s period 4.
Our discussion here, “Miles Periodic Table with Standard Periodic Table reference” recently turned to start page 12. A little review may be in order. I’m not sure exactly how it started, I’d need to reread it. I’m content saying I’d offered my meager programming services and support to a project of Cr6’s choosing, something, he suggested, that might generate a little more charge field interest from people that can use Jupyter Notebook. That and his interest in charge field atoms were in mind for “Miles Periodic Table with Standard Periodic Table reference”. That or I hijacked his thread. We are concerned with refining and sharing our understanding of charge field atomic bonding in and between atoms.
With respect to the proton to proton or slot to slot bonding between the ellipses shown above, “All disks fit together edge to hole, like male and female sockets”. Those bonds between slots, at least within an atom, are all orthogonal, with a 90 degree angle between the two bonded slots’ emission planes.
Within each of the up to 19 elliptical slot stack positions that form an atom, the up to six protons present in each slot bond hole to hole, like parallel or anti-parallel female to female sockets.
In his “Diatomic Hydrogen” (DH) paper, Miles explains how, given a vertically oriented proton spinning left or right, the proton may be accompanied by an electron, orbiting the proton’s top or bottom pole. The electron’s presence blocks a large fraction of charge from entering that proton pole. The result is a low charge field pressure zone near the proton’s opposite pole. This low c.f. zone can be filled with the proton of another, similar ep set, up to six compatible electron-position, and proton-spins.

Since bonding within and between atoms depends on the position and spin of all the epn (electron, proton and neutron) particles within a given slot, our diagrams contain that extra information. Cr6 came up with the Slotlayout (SL) diagram on the left, although the data it originally contained has increased. Following the DH rules, as currently understood, there can be a total of 84 possible combinations for each of the one to six ep-ep slot stack sets. All defined according to +x,+y,+z. The Slotlayout and slot combinations are used by mBuilder, a Jupyter notebook project that can be found at https://github.com/LtAirman/ChargeFieldTopics
ChargeFieldTopics/notebook2/mBuilder.ipynb
mBuilder can display a matplot plot of any single atom, Hydrogen through Thorium (1-90), center. Showing proton spins left or right (blue or red); top or bottom (cyan or black) electrons; and green neutrons – widely orbiting the proton’s low c.f. zone pole. Its nice to be able to dial up any atom as needed. mBuilder can also show matplot plots of any two given slots with any specified proton (or low c.f. zone hole) occupying the other slot’s hole. We’ve yet to figure out which combinations are valid or not.
I often add Autocad atomic and molecular models to my postings, at right, which are not a part of mBuilder, although its likely that the same level of detail can be recreated with another matplitlib plot. The autocad model is comprised of x,y,z aligned circles containing each slot’s epn data as in the SL diagram. One can easily see when a slot has been flipped 180 degrees – an important bonding information detail.
We have also yet to define bonds in which two or more protons or slots can occupy a single proton low c.f. pressure zone.
All in all, I’d say it proves that our project efforts have been cumulative.
I recently mentioned once again re-reading “PERIOD FOUR of the Periodic Table” (PF). There are a few unsettling differences between Miles’ atomic descriptions in “Period Four“ (PF) and our project’s main working assumptions taken from “Diatomic Hydrogen”.
Starting with the fact that DH predated PF by eight months.
And all Miles’ mentions of ionization. What exactly, is ionization? Missing electrons result in missing bonds. I thought ionization levels could possibly indicate energy differential proton size changes, like above proton x or y spin additions or deletions. Another possibility may be that two electrons can orbit a single pole as easily as one; two electrons orbiting a proton pole would result in an even larger lower c.f. pressure zone.
This latest new page absolutely cries out for database progress. Pardon me for saying, you can lead a horse to water but you cannot make him drink. In my case my understanding of T-sql and Neo4j is woefully awful. Please do not assume that I understand your queries at all. I will, however, do my best to follow your instructions.
Cr6, you’re the boss and our database manager. I’m here to help.
http://milesmathis.com/updates.html
NEW PAPER, 9/28/2012. Diatomic Hydrogen. http://milesmathis.com/diatom.pdf My new charge bonding explains this much better than electron sharing. Plus an analysis of spin isomers.
NEW PAPER, 5/15/2013. Period 4 of the Periodic Table. http://milesmathis.com/per4.pdf In this long paper I provide the nuclear diagrams for many elements, including iron, cobalt, nickel, copper, germanium and bromine, showing the mechanical cause of many elemental characteristics. As a bonus, I also show how to diagram a Neodymium magnet.
NEW PAPER, added 4/14/24, The Fractional Quantum Hall Effect. http://milesmathis.com/fraction.pdf As with the Hall Effect, I solve this with the charge field.
NEW PAPER, added 4/20/24, The Rule of Four. http://milesmathis.com/four.pdf Mainstream science is stumped again.
P.S. Changed "two" to "one" - possible number of electrons orbiting a proton pole.
P.P.S. and since the emission plane is the "male" direction, changed "male to male", bonding within a slot to "female to female". Periodic reviews do help sometimes.
.
Last edited by LongtimeAirman on Tue Apr 23, 2024 12:00 pm; edited 2 times in total (Reason for editing : Added P.P.S.)
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Tue Apr 23, 2024 12:43 am
by Chromium6 Tue Apr 23, 2024 12:43 am
Last edited by Chromium6 on Tue Apr 23, 2024 10:03 pm; edited 1 time in total (Reason for editing : Typing on my mobile phone initially.)
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Fri Apr 26, 2024 8:56 am
by Chromium6 Fri Apr 26, 2024 8:56 am
On vacation, will look at this again when I get back.
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Fri Apr 26, 2024 10:33 pm
by LongtimeAirman Fri Apr 26, 2024 10:33 pm

How can we definitively declare this is a valid bond or not?
Cr6 wrote. Great work and analysis LTAM. Yeah we are definitely at a cross roads of "how does this really logically work?". Thinking now on sequencing ...like what binds first? For a molecule, do certain bonds occur first with consequent bonds following? Per C.F. strength?
Airman. Thanks Cr6. They are all open questions. I must admit I spend plenty of time thinking about this stuff, wishing I was a bit smarter. ‘Incremental time’ and ‘sequencing’ sounds like you’re thinking of animations. You’ve also mentioned knowledge graphs a couple of times lately. Is charge field Machine Learning still the goal?
Cr6 wrote. I think we may still be at 80/20 with success. I tried to string together a model. Hopefully we can take it to a gold 3-D model. Graph Databases can show relationships if coded well, but not always the "why" in a clear formula. Miles' papers give some direction for this. Every time an atom binds with another atom, incrementally the C.F. changes. What bonds first and why is still out there..
Airman. Agreed. Unfortunately, for the time being, I'm afraid that without a working set of charge field bonding rules I don’t see how we can graph all possible bonds.
Cr6 wrote. I'll be a sorry punching bag until the models allow good predictability.
Airman. A sorry punching bag? Where the heck did that come from?
Cr6 wrote. Current theory in comparison is pretty awful currently. Like given random X-Y-Z atoms predict the likely molecule they form. Sorry if I'm stating the obvious.
Airman. Even if they can't explain it, mainstream has vast amounts of data that a proper c.f. database could likely make good use of.
Cr6 wrote. Sorry LTAM,
On vacation, will look at this again when I get back.
Airman. Cr6, three sorry statements in a row, you got me worried. I hope everything is Ok.
A vacation sounds good, especially if it does ya some good.
I guess I'll get back to working on c.f. atomic bonding rules.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Sun Apr 28, 2024 5:28 pm
by LongtimeAirman Sun Apr 28, 2024 5:28 pm

Cr6, You posted the on the 3 body problem yesterday which I took to mean you aren’t really on vacation.
https://milesmathis.forumotion.com/t712-the-planetary-orbit-in-netflixs-3-body-problem-is-random-and-chaotic-but-could-it-exist#7626
I’m sure you realize I’m far from familiar or comfortable, and am easily frustrated by the whole process series of graph database operations. I was the punching bag taking a break. Begging to change the subject back to valid or invalid bonds.
Having successfully updated and altered the AtomicMilesMathisOrbitalsDetailAllBonds procedure as per your instructions, I also re-ran your 9 Apr Query for csv file to include any changes.
At AuraDb’s Query section. I detached and deleted the database nodes.
At AuraDb’s import section, Deleted the schema and imported the updated csv file. Recreated the schema with nodes ElementSrc (key ElementSrc), ElementDest (key ElementDest) - including all properties, and the [BINDS_WITH] relationship.
Back at AuraDb’s Query section again. Loaded the 90 elements, 1,244 slots and first 999 rows of 68,910 [BINDS_WITH] relations. Loading the relationships into AuraDb took even longer then it took to enter into them into Neo4j desktop browser – I stopped at a thousand.
Ran ‘MATCH p=()-[r:HAS_SLOT]->() RETURN p’ and obtained the image shown. It looks much the same as the image I posted on 3 April – when using Neo4j, and only a thousand BINDS_WITH relationships.
When I try to run the Cypher query
- Code:
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
WHERE a.ElementSrc = 'aluminium'
RETURN a,b,r
No changes, no records.
Why is that?
All in all I’ve pretty much gone through the entire process about three-four times.
What am I doing wrong? What should I be doing next?
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sun Apr 28, 2024 9:38 pm
by Chromium6 Sun Apr 28, 2024 9:38 pm
You may want to try reformulate your Cypher query. Sorry, this is all still kind of new to me as well. May not have the best insights for root cause or failures to produce output.
Since we are using different file imports it may be that our models are not fully sync'd. My mapping may be slightly different from yours.
I got back from vacation last night. Visited the Grand Canyon which was awesome and impressive.
Was able to rig this query together to show source atoms that share a bond with the ElementDest. I may need to put exclusion filters to prevent them sharing the same (LTAM) Keys-Slots. I think I need to create a single Node as "Element" and then import a new relationship file as "Src - Dest" for valid bonds per Miles. That should allow for a full traversal from Atom1-Rel1-Atom2-Rel2-Atom3-Rel3-etc. We could also attach the "Slots" layouts to each element. I need to add more properties to the relationships so they can be seen better. This is really Alpha at the moment. Also, self-bonds may need to be added as well.
Frankly, I may need to rebuild the whole thing and export it as a Neo4j files so that it can be easily re-imported into AuraDB.
- Code:
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)<-[r1:BINDS_WITH]-(c:ElementSrc)
WHERE a.ElementSrc = 'iron' and c.ElementSrc = 'oxygen'
RETURN a,b,r,r1,c

This query might do it for the Element Node:
- Code:
SELECT DISTINCT X.*
FROM
(
SELECT
[LTAMKeySrc]
-- ,[LTAMKeyDest]
,[AtomicNumberSrc]
-- ,[AtomicNumberDest]
-- ,[SlotDirectionElectronBond]
-- ,[IsValid]
,[ElementSrc]
-- ,[ElementDest]
,[CanBindSrc]
-- ,[CanBindDest]
,[AlphaTypeSrc]
-- ,[AlphaTypeDest]
,[SlotNumberSrc]
-- ,[SlotNumberDest]
,[SlotSpinSrc]
-- ,[SlotSpinDest]
,[AlphaTypeRemainderSrc]
-- ,[AlphaTypeRemainderDest]
,[CarouselAlphaTypeSrc]
-- ,[CarouselAlphaTypeDest]
,[AtomicSymbolSrc]
-- ,[AtomicSymbolDest]
,[SlotOrienSrc]
-- ,[SlotOrienDest]
,[NeutronsSrc]
-- ,[NeutronsDest]
,[ElectronsSrc]
-- ,[ElectronsDest]
,[ProtonsMMSrc]
-- ,[ProtonsMMDest]
,[ProtonsSrc]
-- ,[ProtonsDest]
,[TcountSrc]
-- ,[TcountDest]
,[PXSrc]
,[PYSrc]
,[PZSrc]
,[P2P3Src]
,[P12Src]
,[PESrc]
,[p1xSrc]
,[p1ySrc]
,[p1zSrc]
,[p2xSrc]
,[p2ySrc]
,[p2zSrc]
,[p3xSrc]
,[p3ySrc]
,[p3zSrc]
,[p4xSrc]
,[p4ySrc]
,[p4zSrc]
,[p5xSrc]
,[p5ySrc]
,[p5zSrc]
,[p6xSrc]
,[p6ySrc]
,[p6zSrc]
,[N1N2Src]
,[N1Src]
,[N2Src]
,[N3Src]
,[N4Src]
,[N5Src]
,[N6Src]
,[n1xSrc]
,[n1ySrc]
,[n1zSrc]
,[n2xSrc]
,[n2ySrc]
,[n2zSrc]
,[n3xSrc]
,[n3ySrc]
,[n3zSrc]
,[n4xSrc]
,[n4ySrc]
,[n4zSrc]
,[n5xSrc]
,[n5ySrc]
,[n5zSrc]
,[n6xSrc]
,[n6ySrc]
,[n6zSrc]
,[e1xSrc]
,[e1ySrc]
,[e1zSrc]
,[e2xSrc]
,[e2ySrc]
,[e2zSrc]
,[e3xSrc]
,[e3ySrc]
,[e3zSrc]
,[e4xSrc]
,[e4ySrc]
,[e4zSrc]
,[e5xSrc]
,[e5ySrc]
,[e5zSrc]
,[e6xSrc]
,[e6ySrc]
,[e6zSrc]
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
WHERE CanBindSrc = 1 and IsValid =1
UNION
SELECT
-- ,[LTAMKeySrc]
[LTAMKeyDest]
-- ,[AtomicNumberSrc]
,[AtomicNumberDest]
-- ,[SlotDirectionElectronBond]
-- ,[IsValid]
-- ,[ElementSrc]
,[ElementDest]
-- ,[CanBindSrc]
,[CanBindDest]
-- ,[AlphaTypeSrc]
,[AlphaTypeDest]
-- ,[SlotNumberSrc]
,[SlotNumberDest]
-- ,[SlotSpinSrc]
,[SlotSpinDest]
-- ,[AlphaTypeRemainderSrc]
,[AlphaTypeRemainderDest]
-- ,[CarouselAlphaTypeSrc]
,[CarouselAlphaTypeDest]
-- ,[AtomicSymbolSrc]
,[AtomicSymbolDest]
-- ,[SlotOrienSrc]
,[SlotOrienDest]
-- ,[NeutronsSrc]
,[NeutronsDest]
-- ,[ElectronsSrc]
,[ElectronsDest]
-- ,[ProtonsMMSrc]
,[ProtonsMMDest]
-- ,[ProtonsSrc]
,[ProtonsDest]
-- ,[TcountSrc]
,[TcountDest]
,[PXDest]
,[PYDest]
,[PZDest]
,[P2P3Dest]
,[P12Dest]
,[PEDest]
,[p1xDest]
,[p1yDest]
,[p1zDest]
,[p2xDest]
,[p2yDest]
,[p2zDest]
,[p3xDest]
,[p3yDest]
,[p3zDest]
,[p4xDest]
,[p4yDest]
,[p4zDest]
,[p5xDest]
,[p5yDest]
,[p5zDest]
,[p6xDest]
,[p6yDest]
,[p6zDest]
,[N1N2Dest]
,[N1Dest]
,[N2Dest]
,[N3Dest]
,[N4Dest]
,[N5Dest]
,[N6Dest]
,[n1xDest]
,[n1yDest]
,[n1zDest]
,[n2xDest]
,[n2yDest]
,[n2zDest]
,[n3xDest]
,[n3yDest]
,[n3zDest]
,[n4xDest]
,[n4yDest]
,[n4zDest]
,[n5xDest]
,[n5yDest]
,[n5zDest]
,[n6xDest]
,[n6yDest]
,[n6zDest]
,[e1xDest]
,[e1yDest]
,[e1zDest]
,[e2xDest]
,[e2yDest]
,[e2zDest]
,[e3xDest]
,[e3yDest]
,[e3zDest]
,[e4xDest]
,[e4yDest]
,[e4zDest]
,[e5xDest]
,[e5yDest]
,[e5zDest]
,[e6xDest]
,[e6yDest]
,[e6zDest]
FROM [dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
WHERE CanBindDest = 1 and IsValid =1
) X
Order by 2, SlotNumberSrc
Query for the CAN_BIND relationship:
- Code:
SELECT distinct LTAMKeySrc, LTAMKeyDest, AtomicNumberSrc, AtomicNumberDest , ElementSrc, ElementDest
FROM [dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
WHERE CanBindDest = 1 and CanBindSrc =1 and IsValid =1
order by ElementSrc, ElementDest
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Wed May 01, 2024 10:28 pm
by LongtimeAirman Wed May 01, 2024 10:28 pm

This first image was created in the AuraDB’s ‘Explore’ section which uses Bloom. It looks like the bloom cypher query is equivalent to:
- Code:
MATCH (a:ElementSrc{ElementSrc = 'aluminium'})-[]->(b:ElementDest)
RETURN a,b,r
Cr6, Thanks for coping with my recent near panic. I’m trying to do a better job understanding what I’m doing.
My current schema includes Src and Dest nodes (instead of ElementSrc and ElementDest) for less confusion.

The second image shows the AuraDB Query section output for:
- Code:
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
Still no joy when I try running either of the following two queries:
- Code:
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
WHERE a.ElementSrc = 'aluminium'
RETURN a,b,r
–- Or
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)<- [r1:BINDS_WITH]-(c:ElementSrc)
WHERE a.ElementSrc = 'iron' and c.ElementSrc = 'oxygen'
RETURN a,b,r,r1,c
I just noticed and have yet to look at your latest Create Element Nodes or CAN_BIND relationship queries.
By the way, in those two (previous) T-SQL query outputs, ‘carbon’ is misspelled as ‘cabon’. I see that that error is present in vwAtomicMilesMathisOrbitalsDetailAllBonds.
- Code:
/****** Script for cabon typos ******/
SELECT [Formula]
,[CurrentAtom]
,[ElementSrc]
,[ElementDest]
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
WHERE [ElementSrc] = 'cabon' OR [ElementDest] = 'cabon'
-- returns 56,294 rows
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Thu May 02, 2024 3:08 am
by Chromium6 Thu May 02, 2024 3:08 am
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Fri May 03, 2024 5:21 pm
by LongtimeAirman Fri May 03, 2024 5:21 pm
Cr6 wrote. This query might do it for the Element Node: ... .
Airman. Cr6, the 28 April Element Node query appears to have a problem. The output’s first row contains the imported csv data field names, 83 of them, all ending in ‘Src’.
- Code:
LTAMKeySrc AtomicNumberSrc ElementSrc CanBindSrc AlphaTypeSrc SlotNumberSrc SlotSpinSrc AlphaTypeRemainderSrc CarouselAlphaTypeSrc AtomicSymbolSrc SlotOrienSrc NeutronsSrc ElectronsSrc ProtonsMMSrc ProtonsSrc TcountSrc PXSrc PYSrc PZSrc P2P3Src P12Src PESrc p1xSrc p1ySrc p1zSrc p2xSrc p2ySrc p2zSrc p3xSrc p3ySrc p3zSrc p4xSrc p4ySrc p4zSrc p5xSrc p5ySrc p5zSrc p6xSrc p6ySrc p6zSrc N1N2Src N1Src N2Src N3Src N4Src N5Src N6Src n1xSrc n1ySrc n1zSrc n2xSrc n2ySrc n2zSrc n3xSrc n3ySrc n3zSrc n4xSrc n4ySrc n4zSrc n5xSrc n5ySrc n5zSrc n6xSrc n6ySrc n6zSrc e1xSrc e1ySrc e1zSrc e2xSrc e2ySrc e2zSrc e3xSrc e3ySrc e3zSrc e4xSrc e4ySrc e4zSrc e5xSrc e5ySrc e5zSrc e6xSrc e6ySrc e6zSrc
Or do I need special instructions?
I see the 570 rows identify all the 1-90 atomic bonding extents.
All the data needed can be included with 186 columns, that seems like a lot.
Maybe “Element Node” could be renamed “Element Node Src”, then a third csv import file can be created, “Element Node Dest”?
Cr6 wrote. I think you see what I was trying to get at with linking element to element with Miles' and your linkages for allowed bonds.
Airman. Yes Sir, I think I do. As well as a single-letter change. All in all I’m beginning to appreciate some the complexities involved when working on a better charge field graph model, and queries that can go along with it.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sun May 05, 2024 1:44 am
by Chromium6 Sun May 05, 2024 1:44 am
I've uploaded new files. During the "IMPORT" into AuraDB I found that I can create a Node from the nodes01.csv and just name it "ElementSrc" and then create another new Node and point it to the nodes01.csv and name it "ElementDest". It just creates a reference from the Miles Periodic table in nodes01.csv. The relationships01.csv is from the src-dest query above to show linkages. Dragged the little "+" sign from the ElementSrc to ElementDest and then set the properties relationships. Note, if you want to start from a clean slate in AuraDB, I found deleting everything just doesn't work. Too much old prior work gets mixed in with new file updates. I found that creating a entirely new AuraDB Instance in AuraDBs server admin screen, and then loading new files to it via "IMPORT" is easier than mixing old-new file versions in the same Instance.
I hope this makes sense. I can redo the whole thing and show screenshots for each step if needed.
Files:
New Model file that can be imported-
https://mega.nz/file/z9lA0TyZ#CQQRAaunqLHbH3FXyaB478mMV3Oxn1v7UPybfmu6yNs
nodes01.csv (creates nodes for ElementSrc/ElementDest)
https://mega.nz/file/31kRTYBI#kSPd-ya05u0cs-fV2Mxt5seXUJa83Pumk5fyLrt1HRI
relationships01.csv
https://mega.nz/file/DklmmDTR#tYf4fbZbcuMQsvV0uowL4Faca5K_iMLQUC0z4d2yAsY
I found too that I often have to do a hard browser refresh to see everything with recent changes in a query-explorer window.
This is what I'm seeing now with src-dest:

Last edited by Chromium6 on Sun May 05, 2024 10:48 pm; edited 1 time in total
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Sun May 05, 2024 8:28 pm
by LongtimeAirman Sun May 05, 2024 8:28 pm

Almost, but not quite there yet. My ElementDest node labels are all 0.0 or small real +/- values.
No problem destroying instance01 and obtaining a new instance01. I’d noticed that old nodes were piling up in Aura's Database information section, an instance change is no doubt the correct thing to do.
Imported nodes01.csv and relationships01.csv. Vertical bar separated values were just as easy to read as coma separated values. Both ElementSrc and ElementDest node properties are mapped (select all) from nodes01. Both node types are keyed on their ‘element’ columns.
The Binds_with relationship.
From. Node: ElementSrc. ID: Element. IC column: ElementSrc.
To. Node: ElementDest. ID: Element. IC column: ElementDest.
Our ElementSrc, ElementDest and Binds_with counts are slightly different. Maybe element to same element type is part of the difference?
neo4j_importer_model.json. The New Model json file is new and a nice surprise. AuraDB certainly makes it easy to import csv files. I know that importing a csv file into Neo4j Desktop requires a good deal more effort. I guess you created the json model file so that the database nodes and relationships can be just as easily read by Neo4j Desktop?
Cr6, aside from my small real values its looking good. Having ElementSrc or ElementDest taking on all the same node properties makes sense, and the import seemed to run faster than any previous imports.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sun May 05, 2024 10:39 pm
by Chromium6 Sun May 05, 2024 10:39 pm
For the "Caption-Label" for the ElementDest you can add it by clicking on the gold "ElementDest" label on the right side and select "Element" as the caption. This should do it.

By the way, if you make any changes or leave the AuraDB for a couple of hours...you may need to go back to the "Import" tab and re-import all of the .csv files again. They show up with red question marks. Takes a minute to reimport the .csv files to get back to it live.
May need to look at adding in the Slots as a node for each element. Create a Slots.csv for import as well based on the LTAMKey-Element along with all of the rotations-Proton-Neutron PX-PY layouts.
The counts may be off due to valid links between src-dest (i.e., Slot 17,19 exclusions) this is still kind of beta.
Was also looking at this company in Korea called bitnine which has a commercial Apache Age-postgres database using Cypher queries called AgensGraph. Has a nice interface like AuraDB and has a cypher tutorial. Can do SQL-Json style imports into the AgensGraph database:
AgensGraph
http://bitnine.net/tutorial/tutorial_eng.html
Just an FYI...if you want to manually "traverse" the linkages between Elements you can run this query and right click on an Src or Dest Element and click "expand" to see more linkages. May need to double check this for slot duping/invalid but kind of cool. I might need to double check A-style bonds at higher levels when the carousel is fully occupied with vertical-horizontal slots...like is this really valid? Like it doesn't account for the strength or direction flows without your slot direction values.
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)<-[r1:BINDS_WITH]-(c:ElementSrc)
WHERE b.Element = 'chromium'
RETURN a,b,r, r1, c
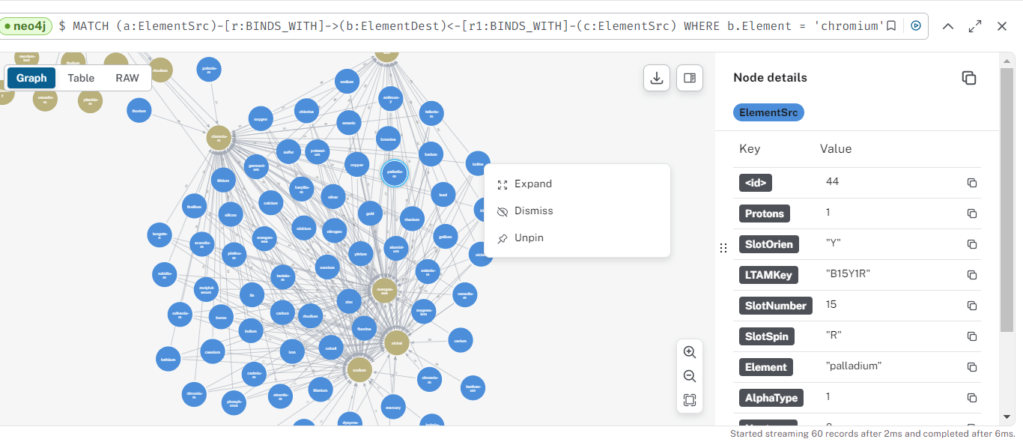
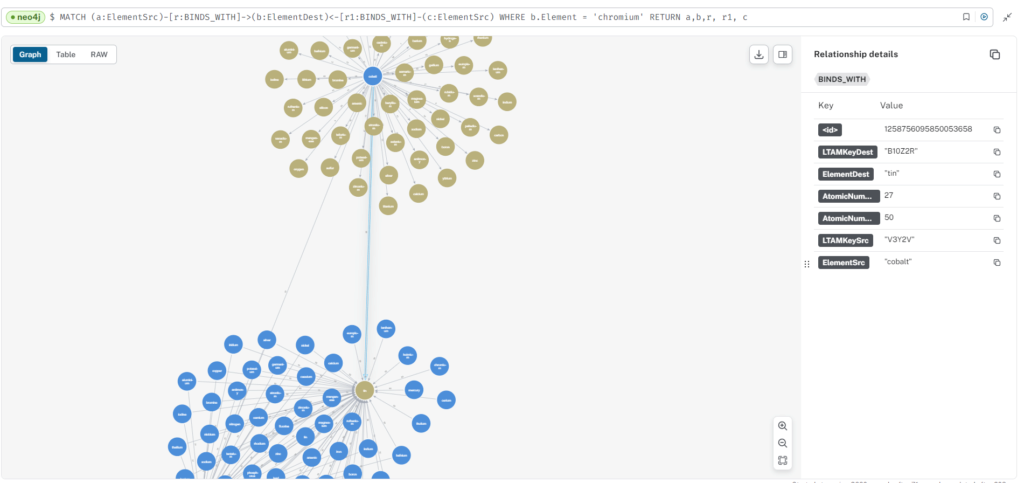
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Mon May 06, 2024 4:34 pm
by LongtimeAirman Mon May 06, 2024 4:34 pm

Cr6, Thank you, clicking on the "ElementDest" label then selecting "Element" corrected the node label.
The image shows the Bloom output of two consecutive queries.
- Code:
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
WHERE a.Element = 'aluminium'
RETURN a,b,r
--And
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)<-[r1:BINDS_WITH]-(c:ElementSrc)
WHERE a.Element = 'iron' and c.Element = 'oxygen'
RETURN a,b,r,r1,c
The scariest moment for me so far was a lost connection – prompting me to identify not the password but the connection url. I backed out and logged out of AuraDB then logged back in as usual.
Cr6 wrote. May need to look at adding in the Slots as a node for each element. Create a Slots.csv for import as well based on the LTAMKey-Element along with all of the rotations-Proton-Neutron PX-PY layouts.
Airman. I think I’m ready, the sooner the better.
Cr6 wrote. Was also looking at this company in Korea called bitnine … AgensGraph
http://bitnine.net/tutorial/tutorial_eng.html .
Airman. Another graph interface? Sounds Ok but I’m still getting used to the three we’ve got. I’m hoping we end up with something compatible with Jupyter Notebook.
Cr6 wrote. Just an FYI...if you want to manually "traverse" the linkages between Elements … .
Airman. Yep, traversals are easy to do and kind of fun. Of course Its too soon to figure out what’s real or not.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Wed May 08, 2024 12:01 am
by Chromium6 Wed May 08, 2024 12:01 am
- Code:
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
WHERE a.Element = 'niobium'
RETURN a,b,r
https://en.wikipedia.org/wiki/Lithium_niobate
Wikipedia wrote:Lithium niobate (LiNbO3) is a synthetic salt consisting of niobium, lithium, and oxygen. Its single crystals are an important material for optical waveguides, mobile phones, piezoelectric sensors, optical modulators and various other linear and non-linear optical applications.[6] Lithium niobate is sometimes referred to by the brand name linobate.[7]
Properties
Lithium niobate is a colorless solid, and it is insoluble in water. It has a trigonal crystal system, which lacks inversion symmetry and displays ferroelectricity, the Pockels effect, the piezoelectric effect, photoelasticity and nonlinear optical polarizability. Lithium niobate has negative uniaxial birefringence which depends slightly on the stoichiometry of the crystal and on temperature. It is transparent for wavelengths between 350 and 5200 nanometers.
Lithium niobate can be doped with magnesium oxide, which increases its resistance to optical damage (also known as photorefractive damage). Other available dopants are iron, zinc, hafnium, copper, gadolinium, erbium, yttrium, manganese and boron.
Growth
A Z-cut, single-crystal lithium-niobate wafer
Single crystals of lithium niobate can be grown using the Czochralski process.[8]
After a crystal is grown, it is sliced into wafers of different orientation. Common orientations are Z-cut, X-cut, Y-cut, and cuts with rotated angles of the previous axes.[9]
Thin films
Thin-film lithium niobate (e.g. for optical wave guides) can be transferred to or grown on sapphire and other substrates, using the smart cut (ion slicing) process[10][11] or MOCVD process.[12] The technology is known as lithium niobate on insulator (LNOI).[13]
Nanoparticles
Nanoparticles of lithium niobate and niobium pentoxide can be produced at low temperature.[14] The complete protocol implies a LiH induced reduction of NbCl5 followed by in situ spontaneous oxidation into low-valence niobium nano-oxides. These niobium oxides are exposed to air atmosphere resulting in pure Nb2O5. Finally, the stable Nb2O5 is converted into lithium niobate LiNbO3 nanoparticles during the controlled hydrolysis of the LiH excess.[15] Spherical nanoparticles of lithium niobate with a diameter of approximately 10 nm can be prepared by impregnating a mesoporous silica matrix with a mixture of an aqueous solution of LiNO3 and NH4NbO(C2O4)2 followed by 10 min heating in an infrared furnace.[16]
Applications
Lithium niobate is used extensively in the telecommunications market, e.g. in mobile telephones and optical modulators.[17] Due to its large electro-mechanical coupling, it is the material of choice for surface acoustic wave devices. For some uses it can be replaced by lithium tantalate, LiTaO3. Other uses are in laser frequency doubling, nonlinear optics, Pockels cells, optical parametric oscillators, Q-switching devices for lasers, other acousto-optic devices, optical switches for gigahertz frequencies, etc. It is an excellent material for manufacture of optical waveguides. It's also used in the making of optical spatial low-pass (anti-aliasing) filters.
[:BINDS_WITH {LTAMKeyDest: "V1Z2V", ElementDest: "lithium", AtomicNumberSrc: 41, AtomicNumberDest: 3, LTAMKeySrc: "T14Y1L", ElementSrc: "niobium"}]
https://en.wikipedia.org/wiki/Pockels_effect (charge flows might explain this effect better!)
LTAM wrote:Airman. Another graph interface? Sounds Ok but I’m still getting used to the three we’ve got. I’m hoping we end up with something compatible with Jupyter Notebook.
Cr6 wrote. Just an FYI...if you want to manually "traverse" the linkages between Elements … .
Airman. Yep, traversals are easy to do and kind of fun. Of course Its too soon to figure out what’s real or not.
Totally understand with sticking with Neo4j which has interface hooks for Jupyter notebooks (which is really cool btw). May need create a "Miles Mathis" chatbot at some point from his papers and the graphs we are making. Could be cool...
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Fri May 10, 2024 10:54 pm
by LongtimeAirman Fri May 10, 2024 10:54 pm

Cr6’s cypher match query returns a graph of a niobium atom (ElementSrc) with 62 [binds_with] links to 62 surrounding atoms (ElementDest nodes). The labels have been changed from ‘Element’ to the atom’s binding slot ‘LTAMkey’ value. That code includes four values: 1. the slot’s alpha type (first and last character values), which can be AA-(TLBR),VV-(TRBL),EE-(TRBR),WW-(TLBL), where TBLR (Top,Bottom,Left,Right) describes the slot’s ep (electron/proton) alpha configuration. 2. the slot number (the second value from the left), an integer 1-19. 3. the slot’s proton emission plane (center value), X, Y or Z. 4. and the number of ep sets or the number of protons in that slot (second value from the right), 1-6.
Airman. Wiki’s description of LiNbO3 does include interesting properties, forming crystals and x y or z aligned crystal wafers used in ‘optical waveguides, mobile phones, piezoelectric sensors, optical modulators and various other linear and non-linear optical applications‘.Cr6 wrote. Was "dorking" around with this today and saw an interesting bind between lithium and niobium.
…
CODE:
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
WHERE a.Element = 'niobium'
RETURN a,b,r
…
[:BINDS_WITH {LTAMKeyDest: "V1Z2V", ElementDest: "lithium", AtomicNumberSrc: 41, AtomicNumberDest: 3, LTAMKeySrc: "T14Y1L", ElementSrc: "niobium"}].
Cr6, you are much more knowledgeable of elemental properties than I am. For practice and discussion sake, I’ll try looking at ‘Lithium and Niobium’.
Did a quick search for LiNb (and NbLi) at https://webbook.nist.gov/chemistry/ and saw that there is no such compound or molecule listed.
Given our current beta state charge field ‘Physics’ graph and database, in order to consider bonds, the first thing we might do is to run your match query. Doing so returns one ElementSrc node – Niobium and 62 binds_with connections to 62 ElementDest nodes. Next I switched the ElementSrc and ElementDest labels from ‘Element’ to ‘LTAMKey’, and show each possible bonding site slot as in the image.
All nodes include bonding slots containing 1-5 protons. All bonds appear to be the female-to-female shared single slot type.
Niobium, is a BR alpha type that may be somewhat arbitrary, yet it constrains what TBRL electron/proton or proton sets it may bind with. For all the 62 ElementDest nodes present, there is one AA alpha type (boron), one EE (sodium), two VV (lithium, beryllium), ten TL and the rest are BR – there are no WW, TR or BL alpha type ElementDest nodes present. The bonds shown appear to me to be consistent with Nb slot15. I confirmed that the Nb ElementSrc and 62 ElementDest bonds were identified in relationhips01.csv.
Next, I went to the source ‘Phyics’ database, scripting an simple T-SQL query to return all molecules containing niobium and lithium.
- Code:
/****** Selecting NbLi molecules ******/
SELECT [Formula]
,[LTAMKeySrc]
,[LTAMKeyDest]
,[ElementSrc]
,[ElementDest]
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
WHERE ([ElementSrc] = 'lithium' AND [ElementDest] = 'niobium') OR ([ElementSrc] = 'niobium' AND [ElementDest] = 'lithium')
Ba2Li3NbN4, Cs2LiNbS4, H25K3Li2Nb5O15, H6LiNbO6, K3Li2Nb5O15, Li2Nb6Cl18, LiNb6Cl19, LiNbF6, LiNbO, LiNbO2, LiNbO3, LiNbS2, LiNbWO6, SrLi2Nb2O7.
The 14 molecules include elements: Ba,Li,Nb,N,Cs,S,H,K,O,CL,F,W,Sr. Which does not compare well with the graph’s 62 elements.
All 14 formulas containing Nb and Li also include at least one additional atom, such as Oxygen, as in LiNbO.
--Here are all the returned rows for LiNbO
Formula LTAMKeySrc LTAMKeyDest ElementSrc ElementDest
LiNbO B7Y4R V1Z2V niobium lithium
LiNbO T11Z1R V1Z2V niobium lithium
LiNbO T9X4L V1Z2V niobium lithium
LiNbO V1Z2V B13Z1L lithium niobium
LiNbO A3Y4A V1Z2V niobium lithium
LiNbO A1Z4A V1Z2V niobium lithium
LiNbO W4Z4W V1Z2V niobium lithium
LiNbO E5Z4E V1Z2V niobium lithium
LiNbO T14Y1L V1Z2V niobium lithium
LiNbO B8X4R V1Z2V niobium lithium
LiNbO V1Z2V B15Y1R lithium niobium
LiNbO V1Z2V T14Y1L ithium niobium
LiNbO B15Y1R V1Z2V niobium lithium
LiNbO B13Z1L V1Z2V niobium lithium
LiNbO V1Z2V T11Z1R lithium niobium
LiNbO B10Z1R V1Z2V niobium lithium
LiNbO V1Z2V B10Z1R lithium niobium
LiNbO T6Y4L V1Z2V niobium lithium
Note that there are 13: Nb binds_with Li, but only 5: Li binds_with Nb. There may not be any actual NbLi bonds present but shouldn’t the count be the same in both directions?
Cr6, it my understanding that the Phyics DB contains all the molecules known to man that you’ve been able to assemble from various sources and searches. I thought the 69K bonds between two atoms were generated from the list of molecules. LiNb is not on the list. In LiNbO, if Li binds_with O and O binds_with Nb, there wouldn’t be any Li/Nb bonds.
Another search at nist.gov for Li*Nb*
https://webbook.nist.gov/cgi/cbook.cgi?Formula=Li*Nb*&NoIon=on&Units=SI
Excluding ions, returns lists:
Li2,Li3,Li4,Li5,Li6,Li7,Li8 and
Nb2,Nb3,Nb4,Nb5,Nb6,Nb7,Nb8,Nb9,Nb10.
Including ions results in even more Nb returns.
No LiNb bonds are included.
Additional searches, Li*O* and Nb*O*, show that both Li and Nb can bond to O.
At which point I might add Li, Nb and O slotlayout diagrams and matplotlib plots and begin trying to assemble LiNbO3.

.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sat May 11, 2024 10:05 am
by Chromium6 Sat May 11, 2024 10:05 am
I should have looked at Lithium Oxide batteries LiO a bit more. LiNb unique bonds apparently don't exist.
Review
Probing the Electrochemical Processes of Niobium Pentoxides (Nb2O5) for High-Rate Lithium-ion Batteries: A Review
https://chemistry-europe.onlinelibrary.wiley.com/doi/full/10.1002/celc.202300581
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sun May 12, 2024 1:53 pm
by Chromium6 Sun May 12, 2024 1:53 pm
As you know, this question is tricky to answer since topology-directions of the atoms-flows are inter-connected. Pressure-volume-temperature-electrical charge-magnetism-gravity, etc. are also in play.
This paper has a few other Niobium Oxides that might need to be added in:
https://www.lehigh.edu/operando/Publications/1991%20Nb-O%20bond%20distance%20by%20Raman.pdf
LiNbO3
SbNbO4
NbOPO4
NaNbO
KNbO
YNbO4
CaNb2O6
AlNbO4
I used a self-join of Miles' Periodic table originally, I may need to setup more rules around "IsValid" for these LiNb type links.
Noticed too that if I drop out the "A3*" style bonds with Niobium...most of the bonding drops out with the other elements. This might need a revisit as well. Low slot numbers tend to "over-represent" a bond with the model-imported files currently. Especially this one:
LTAMKeySrc
"A3Y4A"
Binds:
(:ElementSrc {Protons: 1, SlotOrien: "Y", LTAMKey: "B15Y1R", SlotNumber: 15, SlotSpin: "R", Element: "niobium", AlphaType: 1, Neutrons: 0, AtomicSymbol: "Nb", AtomicNumber: 41, Electrons: "B"})
(:ElementDest {Protons: 2, SlotOrien: "Z", LTAMKey: "V1Z2V", SlotNumber: 1, SlotSpin: "V", Element: "lithium", AlphaType: 1, Neutrons: 2, AtomicSymbol: "Li", AtomicNumber: 3, Electrons: "V"})
[:BINDS_WITH {LTAMKeyDest: "V1Z2V", ElementDest: "lithium", AtomicNumberSrc: 41, AtomicNumberDest: 3, LTAMKeySrc: "T14Y1L", ElementSrc: "niobium"}]
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Tue May 14, 2024 12:35 am
by Chromium6 Tue May 14, 2024 12:35 am
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Tue May 14, 2024 1:35 am
by Chromium6 Tue May 14, 2024 1:35 am

Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Thu May 16, 2024 2:40 am
by Chromium6 Thu May 16, 2024 2:40 am
https://neo4j.com/labs/neodash/2.4/user-guide/extensions/natural-language-queries/
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Fri May 17, 2024 7:27 pm
by LongtimeAirman Fri May 17, 2024 7:27 pm

I used Autocad to create the possible LiNbO3 shown. That’s when I found an internal bonding problem … .
1. Cr6 wrote. I should have looked at Lithium Oxide batteries LiO a bit more. LiNb unique bonds apparently don't exist.
Airman. I guess your point is that Lithium ions do not interact with Niobium Oxide substrates and that may be a big part of the reason why those batteries work best?
When I looked at Lithium and Oxygen slotlayout diagrams a little closer - again, I saw that they both have (TBRL) slot configuration errors. According to the SL diagram, slot1’s +z top end is bound to slot2’s y aligned center. The current Lithium two-slot configuration is, s1 V1Z2V and s2 T2Y1L. V1Z2V’s low charge field pressure zone (low-cf) hole is at the z-aligned s1’s electron vacant center – not at s1’s top or bottom +/-Z ends. S2-T2Y1L’s low-cf hole is at s2’s -y end and not at s2’s center. That combination violates the rule - bonds must occur in a proton low-cf zone. There is no bond between Li s1 and Li s2 as shown. If s1 is a top/bottom (TB) electron configuration type alpha (such as AA (TLBR), VV (TRBL), EE (TRBR), or WW (TLBL)), with an electron vacant center, then slots 2 and 3 must also be TB type alphas. Slots’ 2 and 3 center low-cf zones can bond with both s1’s electron present ends.
There are plenty of elements containing this same error and need to be corrected. The only thing good about this error is that it is internal to the atoms and should not effect (?) atom-to-atom bonding.
Back to Lithium. Currently, Li’s 3 protons occupy two slots. That may also be an error, the three protons may instead occupy a single slot, as in “HOW TO BUILD A NUCLEUS” *. Then any Li s1-TBRL (also including non-TB type alphas such as TRTR, BRBR, BLBL and TLTL) configuration may be valid. That change would definitely effect Li’s ability to bond with other atoms.
2. Cr6 wrote. I used a self-join of Miles' Periodic table originally, I may need to setup more rules around "IsValid" for these LiNb type links.
Noticed too that if I drop out the "A3*" style bonds with Niobium...most of the bonding drops out with the other elements. This might need a revisit as well. Low slot numbers tend to "over-represent" a bond with the model-imported files currently. Especially this one:
LTAMKeySrc "A3Y4A".
Airman. I agree that we may need more “IsValid” rules.
I don’t understand what ‘drop out the "A3*" style bonds’ means. Nb slot3’s code is A3Y4A. Nb Slot3 bonds internally line between Nb slots 1 and 5. Nb slot3 can only bond with another atom (or just a neutron) with a small proton count in Nb’s hook positions, slots 17 and 19.
3. Cr6 wrote. Yeah it may need to take a bit more to align single elements with their associated atoms in terms of flows. I was just trying to base line single atom to atom bonds initially that may be problematic for C.F. for more complex molecules. Granted for molecules that look off in terms of bonds, we'll need to get the logic to identify them and "why". Lower bonds to higher bonds, with exceptions, is a good bet. Thankfully, graph dbs help show this-- mis-alignment than straight tabular x-y datasets.
Airman. Hallelujah brother, aligning atoms in terms of charge flows is definitely a goal. Can’t wait to see whether the database can make corrections or progress any easier.
4. Cr6 wrote. At the end of the day, I guess we are looking for exceptions....like is this "real?"...here's a bond that appears to occur but does it exist in the literature AFAICT: Bismuth, Ytterbium.

Airman. I agree we are looking for exceptions in order to make charge field model changes. We need to verify our charge field model against the current ‘literature’ and data.
Looking at Ytterbium and Bismuth.
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
WHERE a.Element = 'ytterbium' and b.Element = 'bismuth'
RETURN a,b,r
--Returns
Yb B15Y4R binds_with Bi B15Y5R
MATCH (a:ElementSrc)-[r:BINDS_WITH]->(b:ElementDest)
WHERE a.Element = 'bismuth' and b.Element = 'ytterbium'
RETURN a,b,r
--Returns
Bi B15Y5R binds_with Yb B15Y4R
Unfortunately, those two bonds cannot be valid. One reason being there must be orthogonal bonds between each of the identified slot pairs.
I’d say every extent (s10, s11, s12, s13, s14, s15) needs to be a T/B alpha type. The bonds at those slot centers must align with the charge channels through slots s4, s5, s6, s7, s8 and s9. If the slot extents happen to contain an odd number of protons, the low-cf bond sites will of course not be at that slot’s geometric center, but I don’t believe an off-center low-cf hole (say with 2 +y aligned Top ep protons on one side of the slot and 1 -y aligned Bottom ep proton on the other side of the same slot) shouldn't be a big problem, resulting in a spin imbalance for both atoms.
I believe a second requirement for bonding between Yb and Bi is that for any bonds between pairs of orthogonal extents, one of the two atoms must be rotated 90 degrees from the other atom such that when the two extents meet, they will form an x of four spokes similar to a carousel hub. As shown at the left where the Yb has been rotated 90 degrees about the z-axis and positioned above Bi (please pardon the crude copy pasting) such that the resulting Yb x aligned slot15 meets Bi’s y aligned slot14.
I should mention that changing an atom’s orthogonal slot extents to T/B alpha types in order to allow bonding between atoms is this post’s second, major SL diagram (TBRL) identified error and suggested change today. I should start changing the current Cr6Elements csv file. I’ll console myself with the fact that finding and correcting errors also counts as progress.
5. Cr6 wrote. This is something I'm going to try and stand up for the data model: NeoDashboard can translate natural text entries into Cypher queries. Pretty cool.
Airman. On the path to a Charge field Language model? Sounds good.
*
http://milesmathis.com/index.html
314. How to Build a Nucleus without the Strong Force. http://milesmathis.com/stack.html With simple logic and diagrams. 8pp.
P.S. So I started the review to make changes to ElementsPositions.csv and soon realized that Li slot2, T2Y1L of course only has a single proton. My two-slot description of Li was wrong. I still need to review all the atoms and make the general corrections I'd indicated.
Cr6, what's your opinion on making Li a 3 ep single slot atom?
.
Last edited by LongtimeAirman on Sat May 18, 2024 4:26 pm; edited 1 time in total (Reason for editing : Added P.S.)
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sat May 18, 2024 10:57 pm
by Chromium6 Sat May 18, 2024 10:57 pm
The tab delimited file NistMoleculeValidation.csv is below:
https://mega.nz/file/Op9SQYLb#7UMQn_oj6auhS4F8UUfjLxcknXYRdhBK4ifIawH2fGM) <<<--Current File updated again. Had issue with Collation. Now has 4,351 lines.
LTAM wrote:I should mention that changing an atom’s orthogonal slot extents to T/B alpha types in order to allow bonding between atoms is this post’s second, major SL diagram (TBRL) identified error and suggested change today. I should start changing the current Cr6Elements csv file. I’ll console myself with the fact that finding and correcting errors also counts as progress.
No worries...I think we are even still in the exploratory stage. We have something to work with, a model, that can still be tightened up a bit more. We can definitely version these updated files/tables so that we can compare old/new tables if necessary. I kind of expected that non-full carousel molecules outside of single slot types (hydrogen-helium) were going to be problematic within SQL tables. I just wanted to get a Graph DB set up for another look at structures as well as just doing it for building up skills with Neo4j/AuraDB/OrientDB/Cypher/Tinkerpop/etc.
Here are the first few hundred lines. I have created procs in the database to check against NIST similar to pulling ChemSpider data. Can create a new column (IsValidinNIST) in the views with an indication of true/false. Doing self-bonds of the same elements/multi-bonds in a molecule is another need...like (Co2). Was going to include more hydrocarbons in the database once we have structures for this worked out better. Of course the orthogonal bonds between each of the identified slot pairs and topology still needs a revisit...this has ElementSrc-ElementDest type pairs:
----------------
- Code:
Line Content WebbookNistID BondType
1 Chemical Formula Not Found AcBe Mono
2 Chemical Formula Not Found AcH Mono
3 Aluminum, compound with silver (1:1) AgAl Mono
4 Chemical Formula Not Found AgAs Mono
5 AgAu radical AgAu Mono
6 Chemical Formula Not Found AgB Mono
7 Chemical Formula Not Found AgBa Mono
8 Chemical Formula Not Found AgBe Mono
9 AgBi AgBi Mono
10 silver bromide AgBr Mono
11 Chemical Formula Not Found AgC Mono
12 Chemical Formula Not Found AgCa Mono
13 Chemical Formula Not Found AgCd Mono
14 Chemical Formula Not Found AgCe Mono
15 Silver chloride AgCl Mono
16 Chemical Formula Not Found AgCo Mono
17 Chemical Formula Not Found AgCr Mono
18 copper, compound with silver (1:1) AgCu Mono
19 Chemical Formula Not Found AgDy Mono
20 Chemical Formula Not Found AgEr Mono
21 Europium, compound with silver(1:1) AgEu Mono
22 silver fluoride AgF Mono
23 Chemical Formula Not Found AgFe Mono
24 AgGa AgGa Mono
25 Chemical Formula Not Found AgGd Mono
26 Chemical Formula Not Found AgGe Mono
28 Chemical Formula Not Found AgHe Mono
29 Chemical Formula Not Found AgHf Mono
30 Chemical Formula Not Found AgHg Mono
31 silver iodide AgI Mono
32 AgIn AgIn Mono
33 Chemical Formula Not Found AgK Mono
34 Chemical Formula Not Found AgLi Mono
35 Magnesium silver imc AgMg Mono
36 Manganese silver imc AgMn Mono
37 Chemical Formula Not Found AgMo Mono
38 Chemical Formula Not Found AgN Mono
39 Silver, compound with sodium(1:1) AgNa Mono
40 Chemical Formula Not Found AgNb Mono
41 Chemical Formula Not Found AgNi Mono
42 silver (II) oxide AgO Mono
43 Chemical Formula Not Found AgP Mono
44 Chemical Formula Not Found AgPb Mono
45 Chemical Formula Not Found AgPd Mono
46 Chemical Formula Not Found AgPt Mono
47 Chemical Formula Not Found AgRb Mono
48 Chemical Formula Not Found AgRe Mono
49 Chemical Formula Not Found AgRh Mono
50 Chemical Formula Not Found AgRu Mono
51 Chemical Formula Not Found AgS Mono
52 Chemical Formula Not Found AgSb Mono
53 Chemical Formula Not Found AgSc Mono
54 AgSe AgSe Mono
55 Chemical Formula Not Found AgSi Mono
56 Chemical Formula Not Found AgSm Mono
57 Chemical Formula Not Found AgSn Mono
58 Chemical Formula Not Found AgSr Mono
59 Chemical Formula Not Found AgTa Mono
60 AgTe AgTe Mono
61 Chemical Formula Not Found AgTi Mono
62 Chemical Formula Not Found AgTl Mono
63 Chemical Formula Not Found AgTm Mono
64 Chemical Formula Not Found AgV Mono
65 Chemical Formula Not Found AgW Mono
66 Chemical Formula Not Found AgY Mono
67 Chemical Formula Not Found AgZn Mono
68 Chemical Formula Not Found AgZr Mono
69 Aluminum, compound with silver (1:1) AlAg Mono
70 aluminium arsenide AlAs Mono
71 Chemical Formula Not Found AlB Mono
72 Chemical Formula Not Found AlBa Mono
73 Chemical Formula Not Found AlBe Mono
74 Aluminum monobromide AlBr Mono
75 Aluminum carbide AlC Mono
76 Chemical Formula Not Found AlCa Mono
77 Chemical Formula Not Found AlCd Mono
78 Chemical Formula Not Found AlCe Mono
79 Aluminum monochloride AlCl Mono
80 Chemical Formula Not Found AlCo Mono
81 Chemical Formula Not Found AlCr Mono
82 Chemical Formula Not Found AlCu Mono
83 Chemical Formula Not Found AlEu Mono
84 Aluminum monofluoride AlF Mono
85 Chemical Formula Not Found AlFe Mono
86 Chemical Formula Not Found AlGa Mono
87 Chemical Formula Not Found AlGe Mono
89 Chemical Formula Not Found AlHe Mono
90 Chemical Formula Not Found AlHf Mono
91 Aluminum monoiodide AlI Mono
92 Chemical Formula Not Found AlIn Mono
93 Chemical Formula Not Found AlK Mono
94 Chemical Formula Not Found AlLa Mono
95 Chemical Formula Not Found AlLi Mono
96 Chemical Formula Not Found AlMg Mono
97 Aluminum manganese imc AlMn Mono
98 Chemical Formula Not Found AlMo Mono
99 aluminium nitride AlN Mono
100 Chemical Formula Not Found AlNa Mono
101 Chemical Formula Not Found AlNb Mono
102 Chemical Formula Not Found AlNd Mono
103 Aluminum nickel (alni) AlNi Mono
104 Aluminum monoxide AlO Mono
105 aluminium phosphide AlP Mono
106 Chemical Formula Not Found AlPd Mono
107 Chemical Formula Not Found AlRb Mono
108 Chemical Formula Not Found AlRe Mono
109 Chemical Formula Not Found AlRh Mono
110 Chemical Formula Not Found AlRu Mono
111 Aluminum sulfide AlS Mono
113 Chemical Formula Not Found AlSc Mono
114 Aluminum, compound with selenium (1:1) AlSe Mono
115 Aluminum, compound with silicon AlSi Mono
116 Chemical Formula Not Found AlSm Mono
117 Chemical Formula Not Found AlSn Mono
118 Chemical Formula Not Found AlSr Mono
119 Chemical Formula Not Found AlTa Mono
120 Chemical Formula Not Found AlTc Mono
121 Aluminum monotelluride AlTe Mono
122 Aluminum titanium imc AlTi Mono
123 Chemical Formula Not Found AlV Mono
124 Chemical Formula Not Found AlY Mono
125 Chemical Formula Not Found AlZn Mono
126 Chemical Formula Not Found AlZr Mono
127 Chemical Formula Not Found ArBe Mono
128 Chemical Formula Not Found AsAg Mono
129 aluminium arsenide AsAl Mono
130 Chemical Formula Not Found AsB Mono
131 Chemical Formula Not Found AsBa Mono
132 Chemical Formula Not Found AsBe Mono
133 AsBr radical AsBr Mono
134 Chemical Formula Not Found AsC Mono
135 Chemical Formula Not Found AsCa Mono
136 Chemical Formula Not Found AsCd Mono
137 Chemical Formula Not Found AsCe Mono
138 AsCl radical AsCl Mono
139 Chemical Formula Not Found AsCo Mono
140 Chemical Formula Not Found AsCr Mono
141 Chemical Formula Not Found AsCu Mono
142 Chemical Formula Not Found AsDy Mono
143 Chemical Formula Not Found AsEu Mono
144 AsF radical AsF Mono
145 Chemical Formula Not Found AsFe Mono
146 gallium arsenide AsGa Mono
147 Chemical Formula Not Found AsGd Mono
148 Chemical Formula Not Found AsGe Mono
150 Chemical Formula Not Found AsHf Mono
151 Chemical Formula Not Found AsI Mono
152 indium arsenide AsIn Mono
153 Chemical Formula Not Found AsK Mono
154 Chemical Formula Not Found AsLa Mono
155 Chemical Formula Not Found AsLi Mono
156 Chemical Formula Not Found AsMg Mono
157 Chemical Formula Not Found AsMn Mono
158 Chemical Formula Not Found AsMo Mono
159 AsN AsN Mono
160 Chemical Formula Not Found AsNa Mono
161 niobium arsenide AsNb Mono
162 Chemical Formula Not Found AsNd Mono
163 Chemical Formula Not Found AsNi Mono
164 Arsenic monoxide AsO Mono
165 Arsenic monophosphide AsP Mono
166 Chemical Formula Not Found AsPd Mono
167 Chemical Formula Not Found AsPr Mono
168 Chemical Formula Not Found AsRb Mono
169 Chemical Formula Not Found AsRe Mono
170 Chemical Formula Not Found AsRh Mono
171 Chemical Formula Not Found AsRu Mono
172 AsS AsS Mono
173 AsSb AsSb Mono
174 Chemical Formula Not Found AsSc Mono
175 AsSe AsSe Mono
176 Chemical Formula Not Found AsSi Mono
177 Chemical Formula Not Found AsSm Mono
178 Chemical Formula Not Found AsSn Mono
179 Chemical Formula Not Found AsSr Mono
180 Chemical Formula Not Found AsTa Mono
181 Chemical Formula Not Found AsTe Mono
182 Chemical Formula Not Found AsTi Mono
183 Chemical Formula Not Found AsV Mono
184 Chemical Formula Not Found AsY Mono
185 Chemical Formula Not Found AsZn Mono
186 Chemical Formula Not Found AsZr Mono
187 Chemical Formula Not Found AtH Mono
188 Chemical Formula Not Found AtI Mono
189 AgAu radical AuAg Mono
190 Gold, compound with aluminum (1:1) AuAl Mono
191 Chemical Formula Not Found AuAs Mono
192 Gold, compound with boron (1:1) AuB Mono
193 AuBa AuBa Mono
194 AuBe AuBe Mono
195 AuBi AuBi Mono
196 Chemical Formula Not Found AuBr Mono
197 Chemical Formula Not Found AuC Mono
198 AuCa AuCa Mono
199 Chemical Formula Not Found AuCd Mono
200 gold monochloride AuCl Mono
201 Chemical Formula Not Found AuCo Mono
202 Chemical Formula Not Found AuCr Mono
203 Cesium, compound with gold(1:1) AuCs Mono
205 Chemical Formula Not Found AuDy Mono
206 Chemical Formula Not Found AuEr Mono
207 Europium, compound with gold(1:1) AuEu Mono
208 Chemical Formula Not Found AuF Mono
209 AuGa AuGa Mono
210 Chemical Formula Not Found AuGd Mono
211 Germanium, compound with gold(1:1) AuGe Mono
213 Chemical Formula Not Found AuHf Mono
214 Chemical Formula Not Found AuHg Mono
215 gold monoiodide AuI Mono
216 Gold indium imc(auin) AuIn Mono
217 Chemical Formula Not Found AuIr Mono
218 Chemical Formula Not Found AuK Mono
219 Gold, compound with lanthanum (1:1) AuLa Mono
220 Chemical Formula Not Found AuLi Mono
222 Chemical Formula Not Found AuMn Mono
223 Chemical Formula Not Found AuN Mono
224 Gold, compound with sodium(1:1) AuNa Mono
225 Chemical Formula Not Found AuNb Mono
226 Gold, compound with neodymium (1:1) AuNd Mono
227 Gold nickel imc AuNi Mono
228 AuO AuO Mono
229 Chemical Formula Not Found AuP Mono
230 AuPb AuPb Mono
231 Chemical Formula Not Found AuPd Mono
232 Chemical Formula Not Found AuPt Mono
233 Chemical Formula Not Found AuRb Mono
234 AuS AuS Mono
235 Chemical Formula Not Found AuSb Mono
236 Chemical Formula Not Found AuSc Mono
237 AuSe AuSe Mono
238 Gold, compound with silicon (1:1) AuSi Mono
239 Chemical Formula Not Found AuSn Mono
240 AuSr AuSr Mono
241 Chemical Formula Not Found AuTa Mono
242 gold telluride AuTe Mono
243 Chemical Formula Not Found AuTh Mono
244 Chemical Formula Not Found AuTi Mono
245 Chemical Formula Not Found AuTl Mono
246 Chemical Formula Not Found AuV Mono
247 Chemical Formula Not Found AuW Mono
248 Chemical Formula Not Found AuY Mono
249 Gold zinc imc AuZn Mono
250 Chemical Formula Not Found AuZr Mono
251 Chemical Formula Not Found BaAg Mono
252 Chemical Formula Not Found BaAl Mono
253 Chemical Formula Not Found BaAs Mono
254 AuBa BaAu Mono
255 Chemical Formula Not Found BaB Mono
256 Chemical Formula Not Found BaBe Mono
257 Chemical Formula Not Found BaBi Mono
258 Barium monobromide BaBr Mono
259 Chemical Formula Not Found BaC Mono
260 Chemical Formula Not Found BaCa Mono
261 Chemical Formula Not Found BaCd Mono
262 Chemical Formula Not Found BaCe Mono
263 Barium monochloride BaCl Mono
264 Chemical Formula Not Found BaCo Mono
265 Chemical Formula Not Found BaCr Mono
266 Chemical Formula Not Found BaCs Mono
267 Chemical Formula Not Found BaCu Mono
268 Chemical Formula Not Found BaDy Mono
269 Barium monofluoride BaF Mono
270 Chemical Formula Not Found BaFe Mono
271 Chemical Formula Not Found BAg Mono
272 Chemical Formula Not Found BaGa Mono
273 Chemical Formula Not Found BaGd Mono
274 Chemical Formula Not Found BaGe Mono
276 Chemical Formula Not Found BaHe Mono
277 Chemical Formula Not Found BaHf Mono
278 AgAu radical AuAg Mono
279 Chemical Formula Not Found BaHg Mono
280 Chemical Formula Not Found BaHo Mono
281 Barium monoiodide BaI Mono
282 Chemical Formula Not Found BaIn Mono
283 Chemical Formula Not Found BaIr Mono
284 Chemical Formula Not Found BaK Mono
285 Chemical Formula Not Found BAl Mono
286 Chemical Formula Not Found BaLa Mono
287 Chemical Formula Not Found BaLi Mono
288 Chemical Formula Not Found BaMg Mono
289 Chemical Formula Not Found BaMn Mono
290 Chemical Formula Not Found BaMo Mono
291 Chemical Formula Not Found BaN Mono
292 Chemical Formula Not Found BaNa Mono
293 Chemical Formula Not Found BaNb Mono
294 Chemical Formula Not Found BaNd Mono
295 Chemical Formula Not Found BaNi Mono
296 barium oxide, obtained by calcining witherite BaO Mono
297 Chemical Formula Not Found BaOs Mono
298 Chemical Formula Not Found BaP Mono
299 Chemical Formula Not Found BaPb Mono
300 Chemical Formula Not Found BaPd Mono
301 Chemical Formula Not Found BaPo Mono
302 Chemical Formula Not Found BaPt Mono
303 Chemical Formula Not Found BaRa Mono
304 Chemical Formula Not Found BaRb Mono
305 Chemical Formula Not Found BaRe Mono
306 Chemical Formula Not Found BaRh Mono
307 Chemical Formula Not Found BaRu Mono
309 Chemical Formula Not Found BaSb Mono
310 Chemical Formula Not Found BaSc Mono
311 Chemical Formula Not Found BaSe Mono
312 Chemical Formula Not Found BaSi Mono
313 Chemical Formula Not Found BaSm Mono
314 Chemical Formula Not Found BaSn Mono
315 Chemical Formula Not Found BaSr Mono
316 Chemical Formula Not Found BaTa Mono
317 Chemical Formula Not Found BaTe Mono
318 Chemical Formula Not Found BaTi Mono
319 Chemical Formula Not Found BaTl Mono
320 Chemical Formula Not Found BaTm Mono
321 Chemical Formula Not Found BaV Mono
322 Chemical Formula Not Found BaW Mono
323 Chemical Formula Not Found BaY Mono
324 Chemical Formula Not Found BaZn Mono
325 Chemical Formula Not Found BaZr Mono
326 Boron BB Mono
327 Chemical Formula Not Found BBa Mono
328 Chemical Formula Not Found BBe Mono
329 Boron monobromide BBr Mono
Also in AuraDB-Neo4j, I found this query to show everything that links up:
MATCH p=()-->()
RETURN p
Last edited by Chromium6 on Mon May 20, 2024 1:29 am; edited 1 time in total
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Mon May 20, 2024 1:26 am
by Chromium6 Mon May 20, 2024 1:26 am
---------
- Code:
CREATE OR ALTER View [dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
as
SELECT TOP 100 PERCENT ZID,
Formula,
CurrentAtom,
NextAtom1,
NextAtom2,
NextAtom3,
NextAtom4,
NextAtom5,
NextAtom6,
CurrentAtomCount,
NextAtom1Count,
NextAtom2Count,
NextAtom3Count,
NextAtom4Count,
NextAtom5Count,
NextAtom6Count,
LTAMKeySrc,
LTAMKeyDest,
AtomicNumberSrc,
AtomicNumberDest,
SlotDirectionElectronBond,
IsValid,
Replace(TRIM(ElementSrc),'cabon','carbon') as ElementSrc ,
Replace(TRIM(ElementDest),'cabon','carbon') as ElementDest,
CanBindSrc,
CanBindDest,
AlphaTypeSrc,
AlphaTypeDest,
SlotNumberSrc,
SlotNumberDest,
SlotSpinSrc,
SlotSpinDest,
AlphaTypeRemainderSrc,
AlphaTypeRemainderDest,
CarouselAlphaTypeSrc,
CarouselAlphaTypeDest,
AtomicSymbolSrc,
AtomicSymbolDest,
SlotOrienSrc,
SlotOrienDest,
NeutronsSrc,
NeutronsDest,
ElectronsSrc,
ElectronsDest,
ProtonsMMSrc,
ProtonsMMDest,
ProtonsSrc,
ProtonsDest,
[TcountSrc],
[TcountDest],
PXSrc,
PYSrc,
PZSrc,
P2P3Src,
P12Src,
PESrc,
p1xSrc,
p1ySrc,
p1zSrc,
p2xSrc,
p2ySrc,
p2zSrc,
p3xSrc,
p3ySrc,
p3zSrc,
p4xSrc,
p4ySrc,
p4zSrc,
p5xSrc,
p5ySrc,
p5zSrc,
p6xSrc,
p6ySrc,
p6zSrc,
N1N2Src,
N1Src,
N2Src,
N3Src,
N4Src,
N5Src,
N6Src,
n1xSrc,
n1ySrc,
n1zSrc,
n2xSrc,
n2ySrc,
n2zSrc,
n3xSrc,
n3ySrc,
n3zSrc,
n4xSrc,
n4ySrc,
n4zSrc,
n5xSrc,
n5ySrc,
n5zSrc,
n6xSrc,
n6ySrc,
n6zSrc,
e1xSrc,
e1ySrc,
e1zSrc,
e2xSrc,
e2ySrc,
e2zSrc,
e3xSrc,
e3ySrc,
e3zSrc,
e4xSrc,
e4ySrc,
e4zSrc,
e5xSrc,
e5ySrc,
e5zSrc,
e6xSrc,
e6ySrc,
e6zSrc,
PXDest,
PYDest,
PZDest,
P2P3Dest,
P12Dest,
PEDest,
p1xDest,
p1yDest,
p1zDest,
p2xDest,
p2yDest,
p2zDest,
p3xDest,
p3yDest,
p3zDest,
p4xDest,
p4yDest,
p4zDest,
p5xDest,
p5yDest,
p5zDest,
p6xDest,
p6yDest,
p6zDest,
N1N2Dest,
N1Dest,
N2Dest,
N3Dest,
N4Dest,
N5Dest,
N6Dest,
n1xDest,
n1yDest,
n1zDest,
n2xDest,
n2yDest,
n2zDest,
n3xDest,
n3yDest,
n3zDest,
n4xDest,
n4yDest,
n4zDest,
n5xDest,
n5yDest,
n5zDest,
n6xDest,
n6yDest,
n6zDest,
e1xDest,
e1yDest,
e1zDest,
e2xDest,
e2yDest,
e2zDest,
e3xDest,
e3yDest,
e3zDest,
e4xDest,
e4yDest,
e4zDest,
e5xDest,
e5yDest,
e5zDest,
e6xDest,
e6yDest,
e6zDest, -- Id, RepeatText, RepeatCount
N.IsValidinNist
FROM [dbo].[AtomicMilesMathisOrbitalsDetailAllBonds] r
LEFT JOIN dbo.NistMoleculeValidation N on r.AtomicSymbolSrc = N.Element1 and r.AtomicSymbolDest = N.Element2
--CROSS APPLY dbo.NumbersTable2 (1,r.CurrentAtomCount,1) n1
--CROSS APPLY dbo.NumbersTable2 (1,r.NextAtom1Count,1) n2
--CROSS APPLY dbo.NumbersTable2 (1,r.NextAtom2Count,1) n3
--CROSS APPLY dbo.NumbersTable2 (1,r.NextAtom3Count,1) n4
--CROSS APPLY dbo.NumbersTable2 (1,r.NextAtom4Count,1) n5
--CROSS APPLY dbo.NumbersTable2 (1,r.NextAtom5Count,1) n6
--CROSS APPLY dbo.NumbersTable2 (1,r.NextAtom6Count,1) n7
-- WHERE r.Formula = 'Au3Eu7'
-- GROUP BY
-- ZID,Formula,SlotNumberSrc,SlotNumberDest,CurrentAtom,NextAtom1,NextAtom2,NextAtom3,NextAtom4,NextAtom5,NextAtom6,AtomicNumberSrc,AtomicNumberDest,SlotDirectionElectronBond,IsValid,ElementSrc,ElementDest,CanBindSrc,CanBindDest,AlphaTypeSrc,AlphaTypeDest,SlotSpinSrc,SlotSpinDest,AlphaTypeRemainderSrc,AlphaTypeRemainderDest,CarouselAlphaTypeSrc,CarouselAlphaTypeDest,AtomicSymbolSrc,AtomicSymbolDest,SlotOrienSrc,SlotOrienDest,NeutronsSrc,NeutronsDest,ElectronsSrc,ElectronsDest,ProtonsMMSrc,ProtonsMMDest,ProtonsSrc,ProtonsDest,PXSrc,PYSrc,PZSrc,P2P3Src,P12Src,PESrc,p1xSrc,p1ySrc,p1zSrc,p2xSrc,p2ySrc,p2zSrc,p3xSrc,p3ySrc,p3zSrc,p4xSrc,p4ySrc,p4zSrc,p5xSrc,p5ySrc,p5zSrc,p6xSrc,p6ySrc,p6zSrc,N1N2Src,N1Src,N2Src,N3Src,N4Src,N5Src,N6Src,n1xSrc,n1ySrc,n1zSrc,n2xSrc,n2ySrc,n2zSrc,n3xSrc,n3ySrc,n3zSrc,n4xSrc,n4ySrc,n4zSrc,n5xSrc,n5ySrc,n5zSrc,n6xSrc,n6ySrc,n6zSrc,e1xSrc,e1ySrc,e1zSrc,e2xSrc,e2ySrc,e2zSrc,e3xSrc,e3ySrc,e3zSrc,e4xSrc,e4ySrc,e4zSrc,e5xSrc,e5ySrc,e5zSrc,e6xSrc,e6ySrc,e6zSrc,PXDest,PYDest,PZDest,P2P3Dest,P12Dest,PEDest,p1xDest,p1yDest,p1zDest,p2xDest,p2yDest,p2zDest,p3xDest,p3yDest,p3zDest,p4xDest,p4yDest,p4zDest,p5xDest,p5yDest,p5zDest,p6xDest,p6yDest,p6zDest,N1N2Dest,N1Dest,N2Dest,N3Dest,N4Dest,N5Dest,N6Dest,n1xDest,n1yDest,n1zDest,n2xDest,n2yDest,n2zDest,n3xDest,n3yDest,n3zDest,n4xDest,n4yDest,n4zDest,n5xDest,n5yDest,n5zDest,n6xDest,n6yDest,n6zDest,e1xDest,e1yDest,e1zDest,e2xDest,e2yDest,e2zDest,e3xDest,e3yDest,e3zDest,e4xDest,e4yDest,e4zDest,e5xDest,e5yDest,e5zDest,e6xDest,e6yDest,e6zDest
ORDER BY ZID,
Formula,
SlotNumberSrc,
SlotNumberDest,
CurrentAtom,
NextAtom1,
NextAtom2,
NextAtom3,
NextAtom4,
NextAtom5,
NextAtom6,
AtomicNumberSrc,
AtomicNumberDest
GO
Query for finding Non-Valid NIST values. The Binds are at the Element to Element level not necessarily on the Slots. I may need to flag many of these bonds as IsValid=0. Seeing 54,616 of these. I may need to use more update logic for Slot 1-4 for atoms with a full carousel :
SELECT Distinct ElementSrc, ElementDest, LTAMKeySrc, LTAMKeyDest, SlotNumberSrc, SlotNumberDest
FROM [Physics].[dbo].[VwAtomicMilesMathisOrbitalsDetailAllBonds]
WHERE IsValid=1 and CanBindSrc = 1 and CanBindDest=1 and IsValidinNIST =0
order by 1,2,3,4
Here's the new SQL .bak file if you want to restore it. About 3 gigs: https://mega.nz/file/qhsAXApJ#rhbtgSn7Jvv5-3q3cpA_SwE-O7lqok6wsBgVdW9Qvm0
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Mon May 20, 2024 1:30 pm
by LongtimeAirman Mon May 20, 2024 1:30 pm

Running the latest cypher query.
1. When I first tried downloading the csv file, https://mega.nz/file/Op9SQYLb#7UMQn_oj6auhS4F8UUfjLxcknXYRdhBK4ifIawH2fGM)
Mega indicated there was a decryption error – “Enter decryption key. The provided key is invalid. Please check that the key is correct or ask the creator of the link again.”
I unboldened the mega link and tried again and received the same decryption error. For some reason I removed the final character “)” and tried again - the file downloaded successfully.
Verified NistMoleculeValidation_20240520_All.csv contained 1 header row plus 4,351 rows.
2. Downloaded the 6,006,192 KB file, 20240520Physics.bak. Identified it as the Physics backup file and Restored the Physics database to it.
Tried selecting the first 1000 rows of vwAtomicMilesMathisOrbitalsDetailAllBonds and saw the new final column “IsValidInNist”.
Tried executing the “Query for finding Non-Valid NIST values” and saw it returned 54,616 rows. Aside from the fact that that’s a lot of non-valid Nist values ... .
3. Everything looks good.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Tue May 21, 2024 12:40 am
by Chromium6 Tue May 21, 2024 12:40 am
Might be more tricky with lower slot arrangements-bonds (1-3 or 1-6) since they can change depending on "who is bonding". There are probably a lot of Slot 1 and Slot 3 that probably just don't work in the real world (A1-A3). I don't have a lab with all of the elements on a table, so I can't say "does this really bond", per Mathis if it is not found in the literature-publications, and then prove it in the real world with a real experiment? May be an open question for some cases. Orthogonal mapping is probably key here - TBRL - Spin Direction-- your earlier charts as well. TBH, I've always been guilty of trying to cast the net as wide as possible and then refactoring out known issues. From experience, I've always took this approach versus incrementally adding known good -- produces good cases but can't isolate the rules for exclusion as quickly. It is always easier to filter out known issues if included than to expand known good candidates to questionable cases one by one. It is really a question of "how wide do you want to throw the net"? Fisherman are greedy for a good catch.... bad fish are thrown back in the water.
This is a fun query from what we looked at before:
- Code:
SELECT
[ZID]
,[Formula]
,[CurrentAtom]
,[NextAtom1]
,[NextAtom2]
,[NextAtom3]
,[NextAtom4]
,[NextAtom5]
,[NextAtom6]
,[CurrentAtomCount]
,[NextAtom1Count]
,[NextAtom2Count]
,[NextAtom3Count]
,[NextAtom4Count]
,[NextAtom5Count]
,[NextAtom6Count]
,[LTAMKeySrc]
,[LTAMKeyDest]
,[AtomicNumberSrc]
,[AtomicNumberDest]
,[SlotDirectionElectronBond]
,[IsValid]
,[ElementSrc]
,[ElementDest]
,[CanBindSrc]
,[CanBindDest]
,[AlphaTypeSrc]
,[AlphaTypeDest]
,[SlotNumberSrc]
,[SlotNumberDest]
,[SlotSpinSrc]
,[SlotSpinDest]
,[AlphaTypeRemainderSrc]
,[AlphaTypeRemainderDest]
,[CarouselAlphaTypeSrc]
,[CarouselAlphaTypeDest]
,[AtomicSymbolSrc]
,[AtomicSymbolDest]
,[SlotOrienSrc]
,[SlotOrienDest]
,[NeutronsSrc]
,[NeutronsDest]
,[ElectronsSrc]
,[ElectronsDest]
,[ProtonsMMSrc]
,[ProtonsMMDest]
,[ProtonsSrc]
,[ProtonsDest]
,[TcountSrc]
,[TcountDest]
,[PXSrc]
,[PYSrc]
,[PZSrc]
,[P2P3Src]
,[P12Src]
,[PESrc]
,[p1xSrc]
,[p1ySrc]
,[p1zSrc]
,[p2xSrc]
,[p2ySrc]
,[p2zSrc]
,[p3xSrc]
,[p3ySrc]
,[p3zSrc]
,[p4xSrc]
,[p4ySrc]
,[p4zSrc]
,[p5xSrc]
,[p5ySrc]
,[p5zSrc]
,[p6xSrc]
,[p6ySrc]
,[p6zSrc]
,[N1N2Src]
,[N1Src]
,[N2Src]
,[N3Src]
,[N4Src]
,[N5Src]
,[N6Src]
,[n1xSrc]
,[n1ySrc]
,[n1zSrc]
,[n2xSrc]
,[n2ySrc]
,[n2zSrc]
,[n3xSrc]
,[n3ySrc]
,[n3zSrc]
,[n4xSrc]
,[n4ySrc]
,[n4zSrc]
,[n5xSrc]
,[n5ySrc]
,[n5zSrc]
,[n6xSrc]
,[n6ySrc]
,[n6zSrc]
,[e1xSrc]
,[e1ySrc]
,[e1zSrc]
,[e2xSrc]
,[e2ySrc]
,[e2zSrc]
,[e3xSrc]
,[e3ySrc]
,[e3zSrc]
,[e4xSrc]
,[e4ySrc]
,[e4zSrc]
,[e5xSrc]
,[e5ySrc]
,[e5zSrc]
,[e6xSrc]
,[e6ySrc]
,[e6zSrc]
,[PXDest]
,[PYDest]
,[PZDest]
,[P2P3Dest]
,[P12Dest]
,[PEDest]
,[p1xDest]
,[p1yDest]
,[p1zDest]
,[p2xDest]
,[p2yDest]
,[p2zDest]
,[p3xDest]
,[p3yDest]
,[p3zDest]
,[p4xDest]
,[p4yDest]
,[p4zDest]
,[p5xDest]
,[p5yDest]
,[p5zDest]
,[p6xDest]
,[p6yDest]
,[p6zDest]
,[N1N2Dest]
,[N1Dest]
,[N2Dest]
,[N3Dest]
,[N4Dest]
,[N5Dest]
,[N6Dest]
,[n1xDest]
,[n1yDest]
,[n1zDest]
,[n2xDest]
,[n2yDest]
,[n2zDest]
,[n3xDest]
,[n3yDest]
,[n3zDest]
,[n4xDest]
,[n4yDest]
,[n4zDest]
,[n5xDest]
,[n5yDest]
,[n5zDest]
,[n6xDest]
,[n6yDest]
,[n6zDest]
,[e1xDest]
,[e1yDest]
,[e1zDest]
,[e2xDest]
,[e2yDest]
,[e2zDest]
,[e3xDest]
,[e3yDest]
,[e3zDest]
,[e4xDest]
,[e4yDest]
,[e4zDest]
,[e5xDest]
,[e5yDest]
,[e5zDest]
,[e6xDest]
,[e6yDest]
,[e6zDest]
,[IsValidinNist]
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
Where (Formula like '%Li%Nb%O%' or Formula like '%Nb%Li%O%' or Formula like '%O%Nb%Li%' or Formula like '%O%Li%Nb%' or Formula like '%Li%O%Nb%' or Formula like '%Nb%O%Li%')
and [IsValidinNist] = 1 and IsValid=1 and CanBindSrc = 1 and CanBindDest =1
order by ZID, ElementSrc, ElementDest,SlotNumberSrc, SlotNumberDest
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Thu May 23, 2024 8:55 pm
by LongtimeAirman Thu May 23, 2024 8:55 pm

Two charge field atomic models of Carbon from Miles’ ‘The Hydrogen Bond’. Each blue disc represents the emission planes of two protons, and the black discs represent single proton emission planes.
Bonding between protons. The description of the charge field’s atomic model “bonding between protons” used in this project can be found in Miles’ paper, ‘Diatomic Hydrogen’. Bonds can form when protons are accompanied by electrons - as ep pairs or sets. Given a proton with a vertical spin axis, spinning left or right (L R), an electron orbiting either the proton’s top or bottom (T B) pole blocks some of the charge from entering that pole, resulting in a low charge field pressure zone (low-cf) near the proton’s opposite pole. A bond is formed when a neutron or proton or another ep set with complementary allowed electron position (T or B) and proton spin (L or R) direction occupies the first ep’s low-cf zone. The surrounding higher charge pressure keeps the bonded protons together. Up to six ep’s can stack in a vertically aligned single slot, spinning either left or right, or both, although there can be no more than one change of spin direction or no more than a single change of electron pole position within a single stack. Any given stack can have only a single unoccupied low-cf hole, depending on the stack configuration, at either the stack’s top, bottom or middle. A middle stack hole occurs in TB type stacks where the electron position changes and where the proton spin directions may or may not change, and is formed from two opposite and overlapping low-cf zones.
The Slot Layout Diagram - SL. Given the charge field and low/high charge field pressure bonding between protons, when a seventh proton finds its way to the top or bottom low-cf hole of a six ep stack, the combined charge flow of the six aligned protons force the seventh proton to turn its emission plane 90 degrees – orthogonal to the 6 - in order to block some of the charge it receives from the six, thereby forming a second “stack” orthogonal to the first. As each ep is added to a prior stack’s end, atoms appear to grow outward – up, down, left, right, front and back, forming up to 19 occupied slot positions of up to 6 ep’s.
The main vertical up/down channel slots (top to bottom) are (14,4,2,1,3,5,15). There is also the front/back channel (11,8,1,9,13), and left/right channel (10,6,1,7,12); or refer to the directions from center slot 1: +/-x, +/-y, and main vertical column +/-z. The front/back/ left/right arms spin as a single group, the Carousel, around slot 1. Hydrogen has a single proton in its single occupied slot 1. Helium's single slot 1 contains two protons. Hook slot positions: 16,17,18, and 19, are joined to the main up/down column at slots, 2 and 3. Given an atomic number of protons, many atoms have more than one form.

Not allowed (cannot bond) Carbon configurations: s2-A3Y2A, s1-A1Z2A, s3-A3Y2A (SL and plot on the left) and; s2-T2Y1R, s1-A1Z4A, s3-B3Y1L (SL and plot on the right).
I’ve attempted to review, confirm and identify that all adjacent slots within any given atom’s SL diagram is truly “bonded”. I didn’t get very far before seeing that our working assumptions cannot account for every atom. For instance, take possible Carbon, s3-A3Y2A, s1-A1Z2A, s3-A3Y2A (SL and plot on the left). Every slot’s hole is at every slot’s center. By our current rules, s1 cannot bound to s2 or s3. At the same time, neither s2 and/or s3 low-cf hole centers can bond to any s1 proton or hole. Likewise, the three stacks in the second Carbon, (SL and plot on the right), s2-T2Y1R, s1-A1Z4A, s3-B3Y1L, where s1 contains 4 protons and an empty low-cf hole at s1’s center and the two single protons also have empty low-cf holes are also “not bond”.
Cr6 wrote. No worries...I think we are even still in the exploratory stage. We have something to work with, a model, that can still be tightened up a bit more. We can definitely version these updated files/tables so that we can compare old/new tables if necessary.
Airman. I think you're right. Having a model to work with is a good start. Knowing that a ‘database’ offers real advantages when working with model problems is even better. Honestly though, I can’t help but ‘worry’ one thing or another most of the time. I’ve found that its best to treat worries as solvable problems, and to maintain a positive attitude. Like, In considering this post, I realize that we don’t yet have a sufficient set of rules to explain Miles’ Carbon. How might we otherwise account for the two double alphas and top and bottom bonds in Miles’ allowed Carbon slot1?
Cr6 wrote. I don't have a lab with all of the elements on a table, so I can't say "does this really bond", per Mathis if it is not found in the literature-publications, and then prove it in the real world with a real experiment? May be an open question for some cases. Orthogonal mapping is probably key here - TBRL - Spin Direction-- your earlier charts as well. TBH,
Airman. Thanks Cr6. You sound positive. It may seem like grasping at straws, I’ll be thinking about whether it may be possible to add two consecutive ep’s to the center of a single two proton TB alpha, ending up with a BB alpha at s1’s top, butting against a TT alpha at s1’s bottom, and which would then explain how the three Carbon slots might bond.
I’ll also be worrying, wondering whether we're catching or throwing back good fish or bad.
Cr6 wrote. This is a fun query from what we looked at before.
Airman. Yeah, I should have checked [vwAtomicMilesMathisOrbitalsDetailAllBonds] more closely. Your query:
--Returns 281 rows. 8 Formulas: SrLi2Nb2O7, LiNbWO6, LiNbO2, LiNbO3, H6LiNbO6, K3Li2Nb5O15, H25K3Li2Nb5O15, LiNbO.
--With no constraints
--your query returns 4,170,361 rows. 00:06:50. Four million rows indeed. There are 735 rows for Ag2Bi2S3Cl2 alone.
--Where [IsValidinNist] = 1 and IsValid=1 and CanBindSrc = 1 and CanBindDest = 1
--your query returns: 533,304 rows. 00:55. There are 133 rows for Ag2Bi2S3Cl2.
--Where [IsValidinNist] = 0 and IsValid=1 and CanBindSrc = 1 and CanBindDest = 1
--your query Rreturns: 645,775 rows. 1:08. There are 50 rows for Ag2Bi2S3Cl2.
--Where [IsValidinNist] = 1 and IsValid=0 and CanBindSrc = 1 and CanBindDest = 1
--your query returns: 0 rows. 00:01. There are no rows for Ag2Bi2S3Cl2.
http://milesmathis.com/index.html
324. The Hydrogen Bond. http://milesmathis.com/water2.pdf Including a diagram of water. 10pp.
332. Diatomic Hydrogen. http://milesmathis.com/diatom.pdf My new charge bonding explains this much better than electron sharing. Plus an analysis of spin isomers. 9pp.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Fri May 24, 2024 8:37 am
by Chromium6 Fri May 24, 2024 8:37 am
- Code:
SELECT
--ZID, Formula,
--ElementSrc, ElementDest ,
LTAMKeySrc, LTAMKeyDest
--, AtomicNumberSrc,AtomicNumberDest, Count(1) as cnt
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
where
IsValid= 1 and IsValidinNist = 0 and CanBindSrc = 1 and CanBindDest=1
and Case when (AtomicNumberSrc not in (1,2,3,4) and SlotNumberSrc in (1,2) ) or (AtomicNumberDest not in (1,2,3,4) and SlotNumberDest in (1,2) )
then 0
ELSE 1 END =1 --Remove non-available core Carousel Slots
GROUP BY
--ZID, Formula,
-- ElementSrc, ElementDest ,
LTAMKeySrc, LTAMKeyDest
--,AtomicNumberSrc,AtomicNumberDest
order by 1,2
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Tue May 28, 2024 5:27 pm
by LongtimeAirman Tue May 28, 2024 5:27 pm

Once again, please pardon my usual repeated descriptions, thinking while writing and rewriting helps my understanding. Feel free to point out any errors.
This newly revised chart attempts to show most all possible allowed stacks (that is, not including, “unbalanced 6 ep stacks” containing a TB or BBTT bonds not in the middle of the stack), with T or B (cyan or black) electrons and R or L (red or blue) spinning protons. I believe all our current atomic stacks can be found within the eight right side column stacks. Unoccupied low-cf holes are identified by violet circles, near the top or bottom ends. Holes in the middle of the TB stacks are actually two opposite and overlapping top and bottom holes. Every other stack on the right side contains only a single hole near the its top or bottom.
Four new columns containing adjacent (top to bottom) BB and TT alpha configurations in the middle of those stacks can be found in the chart’s four leftmost columns. The 4 ep tall green rectangle shows one of four possible Carbons configurations, tBLBL TRTRb, with low-cf holes above and below slot1. Note that the Carbon alphas have no more than a single change in electron position (from T to B) and no more than a single change in proton spin direction (from R to L or L to R) within that stack. The same basic BBTT configuration also applies to Oxygen, which Miles also includes in The Hydrogen Bond paper. Diagrammed as three z aligned alphas bound to single orthogonal ep protons above and below slot1. The orange rectangle contains one of the four possible Oxygen slot1 configurations tBRBRBR TLTLTLb, again with top and bottom unoccupied holes.
In Diatomic Hydrogen, Miles compares TB and BT, ep to ep configurations, allowing TB and disallowing BT type bonds. Quote.
Why are BT bonds between two single ep’s not allowed, yet the same BT bond is allowed when the two ep’s are a part of four or six ep’s (two or three alphas) in the same slot? There must be a low pressure zone between the BB and TT alphas. The increased current flow between four ep’s vice two, may in fact work to reduce the competing vortices between the two butting electrons present and so allow the BB and TT alphas to get close enough to bond, or maybe I need think more about it.This is why both combinations of top-bottom create bonds and neither combination of bottom-top does. If we have the two electrons nearest eachother—between the protons—the competing vortices keep the atoms from bonding, no matter what way they are spinning [see combinations 7 and 8 in figure 1]. The vortices actually drive the atoms apart. But when we have the electrons opposite, outside the protons, as above, the bond is created no matter the spin directions. The charge field is moving them together regardless, as you see.
Cr6, I ran your latest query,
--LTAMKeys with not valid in NIST but CanBind supposedly and not in Slots (1,2).
It returns 1380 rows.
Which seems to suggest our charge field model predicts more bonds than are known to exist. Adding BBTT alpha bonds to our current bonding repertoire may greatly increase the number of charge field predicted bonds.
http://milesmathis.com/index.html
324. The Hydrogen Bond. http://milesmathis.com/water2.pdf Including a diagram of water. 10pp.
332. Diatomic Hydrogen. http://milesmathis.com/diatom.pdf My new charge bonding explains this much better than electron sharing. Plus an analysis of spin isomers. 9pp.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Wed May 29, 2024 2:49 pm
by Chromium6 Wed May 29, 2024 2:49 pm
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Thu May 30, 2024 5:23 pm
by LongtimeAirman Thu May 30, 2024 5:23 pm

The above is a sideways image of the first 33 of 117 columns of the updated EPNstacks (EPNstacks30May24.csv - not yet posted to github). The BB TT ... BB BT TT configurations added 24 new epn stacks, for a total of 108 select-able epn stacks using the Jupyter Notebook project’s Bonding Widget. The file also includes (1-6), proton, electron and neutron (x,y,z) positions (not shown). A first cut, I haven’t made any code changes, or otherwise verified it yet.
Cr6, I have no idea how you’d go about adding the new bonds to the charge field “Physics” database. Including such stacks as BB TT TT or BB BB TT, and the four spin alternatives, I thought I needed make these latest stacks as clear as possible.
As you know, my current goal is to review and update the 90 Elements-Positions.csv such that all atoms have legitimate internally bound slot layout diagrams. BB TT bonds were definitely needed. I may or may not need another special case bond or two before reaching that goal.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Thu May 30, 2024 11:35 pm
by Chromium6 Thu May 30, 2024 11:35 pm
https://www.microsoft.com/en-us/research/blog/mattersim-a-deep-learning-model-for-materials-under-real-world-conditions/
https://matbench.materialsproject.org/
https://www.microsoft.com/en-us/research/blog/visnet-a-general-molecular-geometry-modeling-framework-for-predicting-molecular-properties-and-simulating-molecular-dynamics/
https://www.microsoft.com/en-us/research/publication/geometric-transformer-with-interatomic-positional-encoding/
https://www.microsoft.com/en-us/research/blog/mattergen-property-guided-materials-design/
PhysicsAssistant: An LLM-Powered Interactive Learning Robot for Physics Lab Investigations
https://arxiv.org/html/2403.18721v1
https://www.microsoft.com/en-us/research/blog/structured-knowledge-from-llms-improves-prompt-learning-for-visual-language-models/
Overcoming the barrier of orbital-free density functional theory for molecular systems using deep learning
https://www.microsoft.com/en-us/research/podcast/abstracts-march-21-2024/
https://en.wikipedia.org/wiki/Orbital-free_density_functional_theory
https://www.microsoft.com/en-us/research/publication/overcoming-the-barrier-of-orbital-free-density-functional-theory-for-molecular-systems-using-deep-learning/
https://www.microsoft.com/en-us/research/podcast/machine-learning-molecular-simulation-and-the-opportunity-for-societal-good-with-chris-bishop-and-max-welling/
They are getting "smaller" but still not "accurate"....because they don't have Miles' structures:
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Sat Jun 01, 2024 4:52 pm
by LongtimeAirman Sat Jun 01, 2024 4:52 pm

24 Bottom-top (BBTT) electron configuration stacks (M, N, P, Q)
Four 4 ep stacks: tBLBL TLTLb, tBRBR TRTRb, tBLBL TRTRb, tBRBR TLTLb,
Eight 5 ep: tBLBL TLTLTLb, tBRBR TRTRTRb, tBLBL TRTRTRb, tBRBR TLTLTLb,
tBLBLBL TLTLb, tBRBRBR TRTRb, tBLBLBL TRTRb, tBRBRBR TLTLb,
Twelve 6 ep: tBLBL TLTLTLTLb, tBRBR TRTRTRTRb, tBLBL TRTRTRTRb, tBRBR TLTLTLTLb,
tBLBLBL TLTLTLb, tBRBRBR TRTRTRb, tBLBLBL TRTRTRb, tBRBRBR TLTLTLb,
tBLBLBLBL TLTLb, tBRBRBRBR TRTRb, tBLBLBLBL TRTRb, tBRBRBRBR TLTLb.
Including all the necessary bond widget proton, electron, neutron and hole coordinates.
EPNStacks30May24 has been posted to github, changed initial P, Q, R, S, to M, N, P, Q, because R was already taken.
Top-bottom (TB) electron configuration type stacks A, V, W, E (TLbtBR, TRbtBL, TLbtBL and TRbtBR) have low-charge pressure ‘holes’ between the protons where the electron position configuration changes within the stack. TB stacks do not form holes outside the stack’s top or bottom ends and therefore cannot provide any proper binding hole ‘foundations’ for slots 2 and 3. True, stack 1 may contribute its proton ends for bonding, the resulting three stack configuration would not agree with the slot layout diagram’s slots 1, 2 and 3.
All our current atomic slot1’s (Hydrogen not included) are (50/50) A or V, TB types. Sorry about that, I see now that those slot assignments were a mistake, made before I had a better understanding.
On the other hand, bottom-top (BBTT) electron configuration stacks as shown above, do form low-cf holes near the top and bottom stack ends which would bond in a proper slots 1 2 and 3 slot layout diagram.
So, for starters, there’s an easy fix, I can simply change all slot1’s to BBTT types. But to which one of the four BT types: M, N, P or Q? Updating Elements-Positions3bt.csv may be all well and good for basic modeling purposes, but given a database, one might expect a range of allowed stacks for any slot. In fact, I do believe there’s no better place to make such range of changes than in a database.
Cr6 I’m still reviewing the slot layout diagrams. How can I best help you add these new bonds?
Thanks for sharing a glimpse of our Mainstream LLM and AI competition. They sure can sure make pretty pictures.
.
Last edited by LongtimeAirman on Sun Jun 02, 2024 8:49 am; edited 1 time in total (Reason for editing : Corrected a couple of 6 ep BBTT config typos.)
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sat Jun 01, 2024 9:00 pm
by Chromium6 Sat Jun 01, 2024 9:00 pm
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Wed Jun 05, 2024 7:55 pm
by LongtimeAirman Wed Jun 05, 2024 7:55 pm

Airman. Cr6, I agree that "we are even still in the exploratory stage"; nevertheless, I’m certain the BBTT epn stack additions are an essential, necessary improvement. Unfortunately, its not unusual for me to rush off to do things before thinking them through. Actually, my original atomic slot1 faux pa epn assignments has made me much more cautious than usual.
1. Hydrogen. H. When a proton is accompanied by an electron, it can be any of 4 (TBLR) types: TL,BR,TR,BL.
2. Helium. He. Maybe the only atom for which the current TB assignment agrees with Miles. All the rest needed changing.
3. Lithium. Li. Quoting. “How to Build a Nucleus”.
4. Berylium. Be. Is described and shown in “How to Build a Nucleus”. We can apply the same analysis to lithium. We have three protons and four neutrons. We stack our three disks, and need four posts to separate them.We can apply the same analysis to lithium. We have three protons and four neutrons. We stack our three disks, and need four posts to separate them.
5. Boron. B. Can be found in “Period Four”.
6. Carbon. C. Can be found in “The Hydrogen Bond”.
7. Nitrogen. N Can be found in “Ammonia”. (Jan 2024).
8. Oxygen. O. Can be found in “Deuterium and Tritium” “The Hydrogen Bond”.With my tweak, we now see the south channel/bond is primary, and why: it is on the nuclear pole of the Nitrogen, matching the charge stream of Nitrogen's seventh proton. And we can see that it is the neutron skewing the whole thing, since the unpictured neutron of Nitrogen is on the upper left side there, excluding the two upper Hydrogens.
9. Fluorine. F. Is mentioned in “Period Four” with similar structure as possible substitute for Boron..
10. Neon. Ne. Can be found in “Deuterium and Tritium” and “The Hydrogen Bond”. 5 alphas, 1 each for slots 1-5.
Quoting. “How to Build a Nucleus”.
Airman. You may have noticed that Neon includes the special case of a single alpha - 2 proton BT configuration. Apparently Neon’s N/S current channel flow somehow allows the BT bond with Ne slot1 top and bottom holes. In which case I suppose I should also add all possible 2 and 3 ep BT stacks to EPNStacks.Our stacking method also explains boron, since we use the one post top and bottom, giving us five protons and six neutrons. As expected, boron 10 is also stable, but it likes to capture an extra neutron to achieve even more symmetry and stability.
With all the smaller elements, this disk stacking is both simple and intuitive. And, as you can see, it continues to keep the E/M field out of the nucleus, even as we go down the periodic table.
2 ep BT: tBLTLb, tBRTRb, tBLTRb, tBRTLb
3 ep BT stacks: tBLBLTLb, tBLTLTLb, tBRBRTRb, tBRTRTRb, tBLBLTRb, tBLTRTRb, tBRBRTLb, tBRTLTLb.
And Neutrons. Neutrons can share a charge channel with a proton, as described in “Deuterium and Tritium”. EPNStacks would also need to identify the alternate neutron positions (where both x and y = 0).
Cr6 wrote. I noticed they are using this networkx based graph library in many cases. https://github.com/graspologic-org/graspologic
Airman. My emphasis. Hu-Ah. Dang tootin. I agree. It is naïve to simply apply statistical algorithms to nodes that contain spatial arrangements.Quoting the Graspologic Overview. A graph, or network, provides a mathematically intuitive representation of data with some sort of relationship between items. For example, a social network can be represented as a graph by considering all participants in the social network as nodes, with connections representing whether each pair of individuals in the network are friends with one another. Naively, one might apply traditional statistical techniques to a graph, which neglects the spatial arrangement of nodes within the network and is not utilizing all of the information present in the graph. In this package, we provide utilities and algorithms designed for the processing and analysis of graphs with specialized graph statistical algorithms.
I see graspologic requires the following packages: networkx, numpy, pandas, scikit-learn, scipy, seaborn.
The Jupyter notebook project already uses the first three. If you deem a graph library is needed, Grasprologic sounds good to me.
http://milesmathis.com/updates.html
NEW PAPER, added 2/6/24, Ammonia. http://milesmathis.com/ammon.pdf With diagrams.
NEW PAPER 7/5/2009. How to Build a Nucleus without a Strong Force, http://milesmathis.com/stack.html with simple postulates and diagrams.
NEW PAPER 11/30/2011. How the Elements are built http://milesmathis.com/nuclear.pdf A mechanical explanation of the periodic table, by diagramming the nuclei.
NEW PAPER, 9/13/2013. Deuterium and Tritium. With diagrams. http://milesmathis.com/deut.pdf Plus analysis of proton-proton reaction in stars.
NEW PAPER 12/20/2011. The Hydrogen bond, http://milesmathis.com/water2.pdf explained without electrons. With a nuclear diagram of water.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Fri Jun 14, 2024 11:49 pm
by LongtimeAirman Fri Jun 14, 2024 11:49 pm

Charge field atomic models for Germanium (Ge, 32) (a,c,d,e), showing 15 slots containing two protons, (2 epn sets) and two hook positions with single proton (epn sets); and along with b. Arlo Emerson’s Molybdenum, (Mo 42) with 15 slots containing four protons (4 epn sets) and two single proton epn sets. Both Ge and Mo may be found in How the Elements are built.
Elements-Positions.csv contains data used to identify 90 different charge field atoms, Hydrogen through Thorium. Each atom can contain up to 19 slots, each slot may contain a stack of up to six sets of electrons, protons and neutrons in valid configurations of electron positions - top or bottom (TB), and proton spins left or right (LR).
EPNStacks.csv is a separate file, intended to contain all possible valid epn stack TBRL configurations along with each stack’s one or two, top and/or bottom low charge field pressure holes. With the latest addition of all the new BT stack configurations I posted to github a few days ago, any given slot may contain one of 144 possible stack configurations.
Sorry, no additional progress to report this last week. I’m still considering changes to Elements-Positions.csv that I’ve mentioned in my last few posts.
1. The first being to replace all the current slot1 TB types configuration assignments with BT types. The reason being that BT slot1 types can bond with slots 2 and 3, while TB slot1 types cannot.
2. All 90 atoms currently contain single atomic solutions. There should be a way to include some atomic variability – such as in identifying a range of possible stacks for any given slot in any given atom. To accomplish that it may be necessary to add two new columns to Elements-Positions, the first containing the slot’s chosen epn stack number (1-144), and the second column could contain a list of alternate epn stacks that that slot's stack could be replaced with.
3. I’d also like to include individual neutrons which can share a charge channel with a proton. That might be accomplished with the addition of a third column, identifying that slot's single neutron special in-line charge channel case.
http://milesmathis.com/updates.html
NEW PAPER 11/30/2011. How the Elements are built http://milesmathis.com/nuclear.pdf A mechanical explanation of the periodic table, by diagramming the nuclei.
P.S. Tried correcting b. in the image and description - failed. Oh well.
.
Last edited by LongtimeAirman on Sat Jun 15, 2024 11:33 am; edited 3 times in total
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sat Jun 15, 2024 11:18 am
by Chromium6 Sat Jun 15, 2024 11:18 am
I've been playing around with this over the last few days. It can take a .pdf file drop and create a Graph in Neo4j out of it.... or several .pdf file drops. Uses ChatGPT 3.5 to parse out .pdfs. Can build a quick chatbot.
https://llm-graph-builder.neo4jlabs.com/
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Thu Jun 20, 2024 1:58 pm
by LongtimeAirman Thu Jun 20, 2024 1:58 pm

Bad BT alpha. There may not be any BT alpha type Heliums, in any case the electrons are colored properly but are in the wrong positions.
Thanks Cr6. Regular posts keep things going. Especially when progress is slow or difficult, they always help focus my thoughts and clarify what needs doing next.
As far as “nice work” goes, thanks again, but it needs improvement. I tried modifying the atomic plot to display a slot1 BT alpha. The result was a cyan or black (T or B) electron near proton 1’s top pole. I removed all code related to neutrons with no change. No excuse, the atomic plot needs reprogramming. Every (1-19) slot in Elements-Positions.csv will point to and be built according to the (1-144) stack selected from EPNStacks.csv. More like the code for the project’s Bonding Widget. Something that might allow for multiple atoms would be nice.
With respect to an atomic ‘graph’, I get the strong impression I’ve missed the boat; sorry I don’t share your appreciation and joy for data. How would we include charge field “Physics” database queries or cyphers into Jupyter Notebook? Please be as specific or obvious as possible when providing help, suggestions and/or directions.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Thu Jun 20, 2024 2:28 pm
by Chromium6 Thu Jun 20, 2024 2:28 pm
Compare the output to the Miles' version in your atom builder.
Here's an atom that popped up "MnAlNI2". They apparently "found" atoms that don't exist yet in literature in some cases. Looks like Microsoft is utilizing this engine for GNNs-Data mining on finding "valid" bonds. I've noticed they use "geometry" a lot in describing the layout.
We have this in the Physics table:
ZID Formula CurrentAtom NextAtom1 NextAtom2
608488 MnAlNi2 Mn Al Ni
--30 rows
SELECT *
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
where Formula like 'MnAlNi2'
and IsValid = 1 and IsValidinNist=1 and CanBindSrc =1 and CanBindDest =1
and SlotNumberSrc > 4 and SlotNumberDest > 4
"Ni₂MnAl is Heusler structured and crystallizes in the cubic Fm̅3m space group. Mn is bonded in a body-centered cubic geometry to eight equivalent Ni atoms. All Mn-Ni bond lengths are 2.50 Å. Ni is bonded in a body-centered cubic geometry to four equivalent Mn and four equivalent Al atoms. All Ni-Al bond lengths are 2.50 Å. Al is bonded in a body-centered cubic geometry to eight equivalent Ni atoms."
----------

Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Fri Jun 21, 2024 1:28 am
by Chromium6 Fri Jun 21, 2024 1:28 am
SELECT *
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds]
where Formula like 'Rb4CO4'
and IsValid = 1 and IsValidinNist=1 and CanBindSrc =1 and CanBindDest =1
"Description (Auto-generated)
Rb₄CO₄ crystallizes in the monoclinic Cm space group. There are three inequivalent Rb¹⁺ sites. In the first Rb¹⁺ site, Rb¹⁺ is bonded to five O²⁻ atoms to form distorted RbO₅ trigonal bipyramids that share corners with two equivalent RbO₅ trigonal bipyramids, an edgeedge with one CO₄ tetrahedra, and a faceface with one CO₄ tetrahedra. There are a spread of Rb-O bond distances ranging from 2.67-2.96 Å. In the second Rb¹⁺ site, Rb¹⁺ is bonded in a 4-coordinate geometry to four O²⁻ atoms. There are a spread of Rb-O bond distances ranging from 2.73-3.24 Å. In the third Rb¹⁺ site, Rb¹⁺ is bonded in a 4-coordinate geometry to four O²⁻ atoms. There are a spread of Rb-O bond distances ranging from 2.66-3.12 Å. C⁴⁺ is bonded to four O²⁻ atoms to form CO₄ tetrahedra that share an edgeedge with one RbO₅ trigonal bipyramid and a faceface with one RbO₅ trigonal bipyramid. There are three shorter (1.44 Å) and one longer (1.45 Å) C-O bond length. There are three inequivalent O²⁻ sites. In the first O²⁻ site, O²⁻ is bonded in a distorted single-bond geometry to four Rb¹⁺ and one C⁴⁺ atom. In the second O²⁻ site, O²⁻ is bonded in a distorted single-bond geometry to four Rb¹⁺ and one C⁴⁺ atom. In the third O²⁻ site, O²⁻ is bonded in a single-bond geometry to five Rb¹⁺ and one C⁴⁺ atom."

Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Fri Jun 21, 2024 11:37 pm
by LongtimeAirman Fri Jun 21, 2024 11:37 pm

I see the 30 row query output for ‘MnAlNi2’. It includes 15 rows of Al-Mn (or Mn-Al) bonds and 15 rows of Al-Ni (or Ni-Al) bonds. The output does not include any bonds between Mn and Ni. Since MnAlNi2 contains as many Ni atoms as there are Mn and Al atoms, it seems unlikely that the query output matches the MnAlNi2 crystal. I could argue that if there are small variations in these atomic stack assignments, there could well be Mn-Ni bonds and that we may indeed be able to find charge field MnAlNi2 molecules.
- Code:
Formula CurrentAtom NextAtom1 NextAtom2 NextAtom3 NextAtom4 NextAtom5 NextAtom6 CurrentAtomCount NextAtom1Count NextAtom2Count NextAtom3Count NextAtom4Count NextAtom5Count NextAtom6Count LTAMKeySrc LTAMKeyDest AtomicNumberSrc AtomicNumberDest SlotDirectionElectronBond IsValid ElementSrc ElementDest
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E B15Y1R 13 25 EEBR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E T14Y2L 13 28 EETL 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T11Z2R E5Z2E 28 13 TREE 1 nickel aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E T11Z1R 13 25 EETR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E B13Z1L 13 25 EEBL 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E B10Z1R 13 25 EEBR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T14Y1L B10Z1R 13 25 TLBR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E T12Z1L 13 25 EETL 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T6Y1L B15Y1R 13 25 TLBR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T6Y1L B10Z1R 13 25 TLBR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E T14Y1L 13 25 EETL 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T14Y1L B15Y1R 13 25 TLBR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T6Y1L T11Z1R 13 25 TLTR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T14Y1L T11Z1R 13 25 TLTR 1 aluminium manganese
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E B15Y2R 13 28 EEBR 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T6Y1L B15Y2R 13 28 TLBR 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E B10Z1R 13 28 EEBR 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E T12Z1L 13 28 EETL 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T14Y1L B15Y2R 13 28 TLBR 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E T11Z2R 13 28 EETR 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL E5Z2E B13Z2L 13 28 EEBL 1 aluminium nickel
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL B13Z2L E5Z2E 28 13 BLEE 1 nickel aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T12Z1L E5Z2E 28 13 TLEE 1 nickel aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T14Y2L B7Y1R 28 13 TLBR 1 nickel aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL B10Z1R E5Z2E 28 13 BREE 1 nickel aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T12Z1L B7Y1R 28 13 TLBR 1 nickel aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL B13Z2L B7Y1R 28 13 BLBR 1 nickel aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T14Y1L B7Y1R 25 13 TLBR 1 manganese aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL T12Z1L B7Y1R 25 13 TLBR 1 manganese aluminium
MnAlNi2 Mn Al Ni NULL NULL NULL NULL 1 1 2 NULL NULL NULL NULL B13Z1L B7Y1R 25 13 BLBR 1 manganese aluminium
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A1Z4A B3Y1R 6 8 AABR 1 carbon oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A3Y4A B3Y1R 37 8 AABR 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A1Z4A A1Z6A 6 8 AAAA 1 carbon oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL B3Y1L B3Y1R 6 8 BLBR 1 carbon oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL T2Y1L T2Y1R 8 6 TLTR 1 oxygen carbon
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A1Z6A T14Y1L 8 37 AATL 1 oxygen rubidium
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A1Z6A B3Y1L 8 6 AABL 1 oxygen carbon
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A1Z6A A1Z4A 8 6 AAAA 1 oxygen carbon
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A1Z4A T2Y1L 6 8 AATL 1 carbon oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL V2Y4V B3Y1R 37 8 VVBR 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A3Y4A T2Y1L 37 8 AATL 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL V2Y4V T2Y1L 37 8 VVTL 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL E5Z4E B3Y1R 37 8 EEBR 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL E5Z4E T2Y1L 37 8 EETL 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL E5Z4E A1Z6A 37 8 EEAA 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A3Y4A A1Z6A 37 8 AAAA 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL V2Y4V A1Z6A 37 8 VVAA 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL T14Y1L B3Y1R 37 8 TLBR 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL T6Y4L B3Y1R 37 8 TLBR 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL T9X4L B3Y1R 37 8 TLBR 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL B8X4R A1Z6A 37 8 BRAA 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL T9X4L A1Z6A 37 8 TLAA 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL B7Y4R A1Z6A 37 8 BRAA 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL T6Y4L A1Z6A 37 8 TLAA 1 rubidium oxygen
Rb4CO4 Rb C O NULL NULL NULL NULL 4 1 4 NULL NULL NULL NULL A1Z6A T2Y1R 8 6 AATR 1 oxygen carbon

Likewise, I see a 25 row SQL query output for ‘Rb4CO4’. Eight rows of C-O (or O-C) bonds, and 17 rows of Rb-O (or single O-Rb) bonds. There are no Rb-C (or C-Rb) bonds. Again, the SQL output doesn’t appear to agree with the Materials Explorer crystal. In this particular molecule, we might expect multiple C or O atoms to bond at some non-orthogonal angle with Rb, but we don’t have those angled bonding rules yet.
Or your two molecular posts might be a gentle reminder that any atomic reprogramming on my part should include molecules identified by the SQL outputs. I would agree, that’s definitely a goal.
What am I missing?
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Mon Jun 24, 2024 9:54 am
by Chromium6 Mon Jun 24, 2024 9:54 am
I think you see it. I may need to just look at how the atoms chain together via allowed bonds. I think you covered it. Angled bonds is tricky.
I just thought these models might be useful to see "bonds" per the literature.
If you drop off "IsValidNist" it produces results:
SELECT A.*
FROM [Physics].[dbo].[vwAtomicMilesMathisOrbitalsDetailAllBonds] A
where Formula = 'MnAlNi2'
and CanBindSrc = 1 and CanBindDest = 1
and (ElementDest = 'manganese' and ElementSrc = 'nickel'
or ElementSrc = 'nickel' and ElementDest = 'manganese')
and IsValid = 1
--and IsValidinNist=1
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Tue Jun 25, 2024 6:21 pm
by LongtimeAirman Tue Jun 25, 2024 6:21 pm

“Correctly” showing a Carbon slot1 BBTT alpha type as read from Elements-Positions4.csv. Slot1:(P: tBLBLTRTRb), slot2:(TRb), slot3:(tBL). The other BT types are: M. tBLTLb. C. tBRTRb. Q. tBRTLb. Single proton epn stacks in slots 2 and 3 are now “bound" occupying slot1’s top and bottom relatively low charge field pressure ‘holes’ in close facsimile to a well-positioned slot layout diagram's slots 1 2 and 3.
Cr6 wrote. I think you see it. I may need to just look at how the atoms chain together via allowed bonds. I think you covered it. Angled bonds is tricky.
I just thought these models might be useful to see "bonds" per the literature.
Airman. They’re certainly useful alright. I’m willing to accept the Materials Explorer images, as well as your 30 May molecular images as reliably good faith mainstream efforts to portray ‘accurately measured’ molecular configurations. At some point I would no doubt register in order to see the 3d interactive Molecular Explorer renderings.
I suppose we’d need to add each individual atom’s size, configuration, emissions and channels before we could possibly understand angled bonds.
My previous post’s Bad BT Helium “electrons at the wrong proton pole position” problem was caused by the “Write particle coordinates to the data frame” cell, which saves the unchanged default or user selected dimension based particle positions. It needs the new BT alpha types added. Till then I’ll just bypass it.
First, I needed to add the BT types to the “Identify all atomic particle locations” cell. I then decided to compress the cell’s code somewhat by adding 4, each 5 line functions: spinLeft, spinRight, botElectron, topElectron - according to the alpha type and particle (x,y,z) coordinates – 34 1-line calls opposed to three times 34 previous lines of code. A total of 54 lines added to remove 102.
The neutrons look like they need work. I have a hard time believing that one or more neutrons could share an orbit in the space between protons 1 and 2. Neutrons sharing the same charge channel with the stack’s protons anywhere along the stack makes sense. In-line protons and neutrons remains on my to-do list.
Next. Currently, in order to change any atom’s slot layout configuration, one must make all (x,y,z) particle position changes within Elements-Positions. Let those existing particle positions be the atoms’ default SL values. My next step is to try reading and assembling each slot’s particle positions from the stacks contained in EPNStacks, and that any given slot in Elements-Position can point too. One may then change or randomize any atomic slot configuration with a single numerical values identifying the desired slot configuration (1-144 (not counting custom neutron configurations)). It will make varying an atom’s SL much easier.
Before switching to the new 1-144 EPNStacks, I’ll also need to reprogram the project’s bonding widget to read and plot the new possible orthogonal bonding configurations.
I'll check out the "IsValidNist" I just noticed in your last post.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Sun Jun 30, 2024 11:42 pm
by Chromium6 Sun Jun 30, 2024 11:42 pm
Just wanted to introduce this OPENUSD from Nvidia might be worth looking at long-term and diagramming the C.F.:
https://www.nvidia.com/en-us/omniverse/
https://changelog.com/practicalai/209#t=894
https://developer.nvidia.com/blog/building-simulation-ready-usd-3d-assets-in-nvidia-omniverse/
https://developer.nvidia.com/blog/new-video-series-what-developers-need-to-know-about-universal-scene-description/?ncid=cont-412344-vt49
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Mon Jul 01, 2024 8:27 pm
by Chromium6 Mon Jul 01, 2024 8:27 pm
https://openusd.org/docs/index.html
https://openusd.org/release/tut_usd_tutorials.html
https://github.com/PixarAnimationStudios/OpenUSD
https://openusd.org/release/api/usd_physics_page_front.html
https://github.com/usd-wg/assets/tree/main/full_assets/StandardShaderBall
Like with Neo4j they have something cooking with LLMs. Neo4j had a decent wrap for creating Cypher with an LLM...looks like OpenUSD has an LLM that allows forms of generation: https://nvidia.github.io/TensorRT-LLM/index.html
Algos they have for NVIDIA...probably related to the stock price with creatable USD "worlds".
https://github.com/NVIDIA/TensorRT-LLM/tree/main/examples
https://github.com/NVIDIA/DeepLearningExamples
Sorry to over-dump this stuff. Nvidia demo of the physics model:
Chromium6- Posts : 818
Join date : 2019-11-29
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Tue Jul 02, 2024 9:32 pm
by LongtimeAirman Tue Jul 02, 2024 9:32 pm

Bad Carbon.
Sharing is caring, reporting slow progress with a quick confusing status. The 3 (of 19) occupied Carbon slots identified in Elements-Positions.csv (and in output 353), from slotsetA_df are: s1: P1Z4P, (tBLBLTRTRb) stack #59; s2: tBR, stack #2; and s3: TLb, stack #1. Those values do not agree with the plot, which was created from the three ‘StackNum’ values 60.0, 1.0 and 2.0, from EPNStacks.csv (dfepn) and shown in output 358. S1: Q1Z4P, tBRBRTLTLb stack #60; s2: TLb, stack #1; and s3: tBR stack #2. While there are 1-144 stacks, the dfepn locations point to the index values, which include 0 to 143; 60-1, 1-1 and 2-1. Slots 2 and 3 are not yet aligned to the y-axis.
P.S. Corrected typo, epn stack #60 from 'Q1Z4P' to 'QiZ4Q'. Added 2 's's for dfepn slots s2 and s3.
.
Last edited by LongtimeAirman on Wed Jul 03, 2024 12:07 pm; edited 2 times in total (Reason for editing : Added P.S.)
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Mon Jul 08, 2024 4:02 pm
by LongtimeAirman Mon Jul 08, 2024 4:02 pm

Good Neon plot. Bad Neon SL (SlotLayout diagram). The plot shows electron, proton and neutron (epn) coordinate positions read from (1-144) stacks listed in EPNStacks.cv (dfepn). All alpha stacks are oriented as intended. Note that BT alpha stacks occupy slots 1, 4 and 5. TB alphas occupy stacks 2 and 3. Proton-to-proton bonding occurs within any single alpha. The bonds between s1, s2 and s4 (or s1, s3, and s5) seem to be a single shared low presure zone alpha-alpha bonding. The neutrons are currently along for the ride, details yet to be specified, part of each and every epn stack.
First Carbon, now Neon. Both atoms are properly assembled according to selected epn stack numbers. Moving on, of course I made changes to see a(n) SL but have no idea how the SL is identifying epn stack numbers 5, 6, 7, 8 instead of 13, 9, 10, 14, 15 as per the info I included at the bottom right.
For a quick reference, here are our current Twelve ‘allowed’ epn stack types.
All 144 stacks of 1-6 epn sets listed fall into one of these 12 types.
t, b, bt, t b are the stacks’ low charge field pressure “holes”.
Four ‘constant’, single electron position and single proton spin type stacks:
TRTRb, tBRBR, tBLBL, TLTLb.
The remaining 8 stack types contain one or both: 1. an electron configuration change (B and T) or (T and B), and 2. a proton spin change (R and L) or (L and R).
Four TB alpha types (A, V, E, W):
A. TLbtBR. L and R spinning protons, T and B electrons.
V. TRbtBL. R and L spinning protons, T and B electrons.
E. TRbtBR. R spinning protons, T and B electrons.
W. TLbtBL. L spinning protons, T and B electrons.
Four BT alpha types (M, C, P, Q):
M. tBLTLb. L spinning protons, B and T electrons.
C. tBRTRb. R spinning protons, B and T electrons.
P. tBLTRb. L and R spinning protons, B and T electrons.
Q. tBRTLb. R and L spinning protons, B and T electrons.
Cr6, I reviewed a few of your animation links. Good references, I'll need to get back to it in the future, its more than I can cope with for now.
Your patience is appreciated.
P.S. 1. Replaced the image, reordering the bottom right slot data to s5, s3, s1, s2, s4 to match the top to bottom order of the plot and SL. 2. Corrected V to TRbtBL. 3. Added holes TRTRb, tBRBR, tBLBL, TLTLb.
.
Last edited by LongtimeAirman on Wed Jul 10, 2024 12:00 pm; edited 3 times in total (Reason for editing : Added P.S.)
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by LongtimeAirman Thu Jul 11, 2024 4:55 pm
by LongtimeAirman Thu Jul 11, 2024 4:55 pm

The SL now properly indicates Neon’s ‘selected’ stack configurations. Agreeing with the plot and TRBL electron position and proton spin details shown. Some clear thinking was needed to add new pointers between slotsetA_df and dfepn.
Last time Neon’s plot and data showed stacks (s1-s5):13, 9, 10, 14, 15. Here, keeping s1-s5 TB and BT alpha type slots the same, I arbitrarily reassigned Neon’s spins, by changing Elements-Positions.csv, “StackNum” column contents to: 16, 12, 11, 15, 14. No mus, no fuss, easy to do. Adding alternative slot lists allowing for more considered atomic variations is a logical next step.
Still need to correct the “Write particle coordinates to the data frame” cell, as well as add the new BT stacks to the “Bonding Widget”.
But first, my current project working file, mBuilder15Jun.ipynb, contains many changes not yet included in mBuilder at github. Before I start updating, I’ll go through all the slots of all the atoms to ensure Elements-Positions.csv contains valid “StackNum” column slot assignments and that EPNStacks.csv is as error free as possible.
Cr6, it occurs to me that, given the proper SQL methods and tools, and the “Physics” database, you may be well ahead of me. Is there anything about these latest project changes that impact the database? Please feel free to share your thoughts, druthers or whatevers.
.
LongtimeAirman- Admin
- Posts : 2078
Join date : 2014-08-10
Chromium6 likes this post
Re: Miles Periodic Table with Standard Periodic Table reference
![]() by Chromium6 Thu Jul 11, 2024 8:54 pm
by Chromium6 Thu Jul 11, 2024 8:54 pm
I may need to break investigations into these 3-D style tools off on another Topic thread that could be used at some point for diagramming the Periodic Table via Miles' publications. Just don't want a big "mix" of over-lapping tools to develop (like it already has...
Was looking at Khronos-glTF that can integrate with OpenUSD: https://github.khronos.org/glTF-Sample-Viewer-Release/
One thing that popped out that got me thinking about how molecules form is their description of PBR:
https://github.com/KhronosGroup/glTF-Tutorials/blob/main/PBR/README.md (mentions Fresnel Equations and Snell's Law for properties of "reflection")
https://www.khronos.org/blog/building-bridges-in-3d-aousd-and-khronos-collaborate-on-openusd-and-gltf-interoperability
This got me trying to conceive of how photon spins create reflections both on screen and real life (and in the human brain):
glTF wrote:Physically-Based Rendering: From Theory to glTF
Mohamad Moneimne, University of Pennsylvania
What is PBR?
Physically-Based Rendering (PBR) refers to techniques that attempt to simulate light in order to render photorealistic images. As indicated by the name, these techniques focus on our understanding of physics to model how light interacts with surfaces that have different physical properties. Since these interactions happen on a very fine level, PBR techniques often use statistical models to add realism and complexity to renders.
PBR has been around for several years now, but was initially too computationally expensive to be a viable option for real-time applications. However, with the continuous advancement of computing power, it has increasingly become an industry standard in real-time graphics. In fact, much of the real-time software we see today such as Unreal Engine 4, Unity 5, Frostbite, and many others use physically-based rendering techniques to provide their users with the ability to create highly realistic 3D scenes.
The goal of this article is to provide some intuition behind PBR theory and cover a bit of the mathematical foundation before discussing the relationship between PBR and glTF.
From Marmoset Toolbag Tutorials: Physically-Based Rendering, And You Can Too!
, by Joe "Earthquake" Wilson
How do we model light-object interactions in PBR?
The physics law most central to PBR is the law of conservation of energy. This law states that the total amount of energy within an isolated system remains constant, but how does this relate to rendering? In PBR, radiance is the energy that is conserved, meaning the amount of incoming light at any point in the scene is equal to the sum of the reflected, transmitted, and absorbed light at that point.
Within any environment, it is easy to see several examples of complicated surfaces that seem to interact with light differently. For example, mirrors reflect perfect images, plastics are shiny, and chalkboards are matte. All of these unique properties can be modeled by considering general mathematical functions called Bidirectional Scattering Distribution Functions (BSDFs). These functions describe how light scatters upon contact with a surface based on the properties that surface holds. More specifically, they follow a statistical model to tell the user how likely the incident light is scattered in a specific outgoing direction.
BSDF sounds like a very complicated term for what it actually means, so let’s break it up and explain its parts...
Bidirectional refers to the notion that at any point on a surface, light comes in and light goes out.
Scatter describes that light coming from one direction onto a surface can end up splitting into a range of directions. For example, light can either scatter by being reflected from or transmitted through the surface in certain directions.
Finally, the details for how light scatters can be described using distribution functions, which entail how light is likely to be distributed in certain directions based on the physical properties of the surface. This can be anything from an equal scatter in all directions to a perfect reflection in a single direction.
To help better understand the kinds of BSDFs that occur, we can consider two general types...
BRDFs (Bidirectional Reflectance Distribution Functions) specifically correspond to BSDFs that describe how light is reflected from a surface. This reflected light refers to the colors we see coming directly from a surface. At this point, it is normal to ask something along the lines of the following: If I shine a white light at a banana, why does it appear yellow instead of white? This is because not all light is just reflected from a surface. While surfaces reflect light of certain colors (wavelengths), they absorb or transmit the remaining energy. For bananas, wavelengths in the yellow spectrum are mainly reflected while other wavelengths are absorbed.
BTDFs (Bidirectional Transmittance Distribution Functions) specifically correspond to BSDFs that describe how light is transmitted through a surface. This can be seen in examples such as glass and plastics where we can see light that has traveled through the surface.
There exist other types of density functions that account for effects such as subsurface scattering (the effect in which light enters a material and bounces around before exiting again in some other position and direction).
What are the reflection models?
There are four general surface types with reflection distribution functions (BRDFs) that describe the probability that light scatters in all directions:
Diffuse – surfaces that scatter light equally in all directions, e.g., even color of a chalkboard
Glossy specular – surfaces that scatter light preferentially in a set of reflected directions and show blurry reflections, e.g., specular highlights on plastic
Perfect specular – surfaces that scatter light in a single outgoing direction such that the angle of incident light is equal to the outgoing light with respect to the surface normal, e.g., perfect reflection of mirrors
Retro-reflective – surfaces that scatter light primarily back along the incident direction of the light source, e.g., specular highlights on velvet
How much light is reflected and transmitted?
It is important for physically-based renderers to know how much light is reflected or transmitted on a surface. It is a combination of these effects that describe substances such as honey and stained glass that both have color and can be seen through.
These amounts are directly related to each other and described by the Fresnel equations. The equations are described for two types of media, dielectrics and conductors.
Chromium6- Posts : 818
Join date : 2019-11-29
Sponsored content
Page 12 of 13 • 1, 2, 3 ... , 11, 12, 13 ![]()
» Cr6's Periodic Table
» RSC's Periodic Table for Comparison
» Periodic Table of Toxic Elements
» Happy sesquicentennial to the periodic table of the elements